Advanced One-Pot Synthesis of 2-Trifluoromethyl Quinazolinones for Scalable Pharmaceutical Production
Advanced One-Pot Synthesis of 2-Trifluoromethyl Quinazolinones for Scalable Pharmaceutical Production
The pharmaceutical and agrochemical industries continuously seek robust synthetic routes for heterocyclic scaffolds that offer enhanced metabolic stability and bioavailability. Patent CN112480015B introduces a significant breakthrough in this domain by disclosing a highly efficient multi-component one-pot method for synthesizing 2-trifluoromethyl substituted quinazolinones. This class of compounds is pivotal in medicinal chemistry, serving as the core structure for numerous bioactive molecules ranging from antifungals to anticancer agents. The strategic incorporation of the trifluoromethyl group is particularly valuable, as it dramatically improves the lipophilicity and metabolic resistance of the parent molecule without significantly increasing steric bulk. For R&D directors and process chemists, this patent represents a critical advancement, offering a streamlined pathway to access these high-value intermediates with superior operational simplicity compared to traditional multi-step sequences.
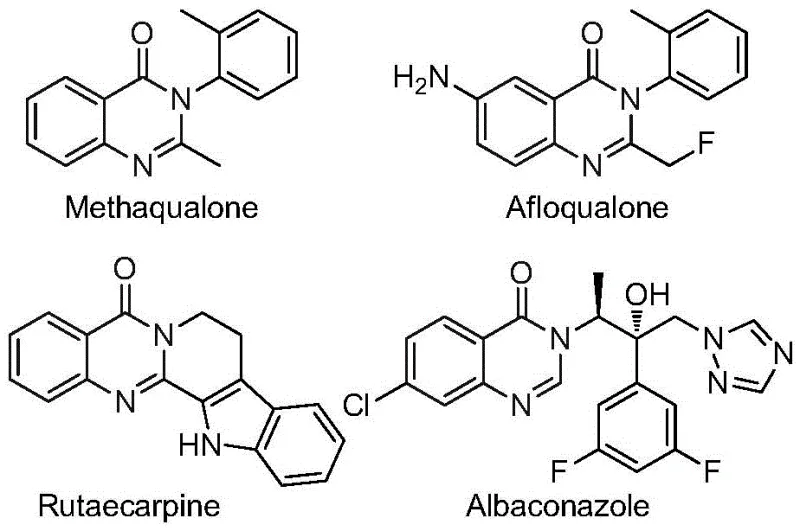
The significance of the quinazolinone scaffold cannot be overstated, as evidenced by its presence in established drugs such as Methaqualone and Afloqualone, shown in the structural overview above. However, accessing specifically 2-trifluoromethyl substituted variants has historically been challenging due to the harsh conditions required for introducing the fluorinated moiety. The disclosed technology addresses this gap by leveraging a palladium-catalyzed carbonylation cascade that operates under relatively mild thermal conditions. By utilizing readily available nitro compounds and trifluoroethylimidoyl chlorides as starting materials, the method bypasses the need for pre-functionalized amines or expensive acid chlorides. This approach not only simplifies the supply chain for raw materials but also reduces the overall step count, making it an attractive candidate for the commercial scale-up of complex pharmaceutical intermediates.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of quinazolinone derivatives has relied on methodologies that impose significant burdens on process safety and cost efficiency. Traditional routes often involve the use of high-pressure carbon monoxide gas, which necessitates specialized autoclave equipment and rigorous safety protocols, thereby inflating capital expenditure for manufacturing facilities. Furthermore, many existing protocols utilize precious metal catalysts like ruthenium or platinum, which are not only costly but can leave trace metal impurities that are difficult to remove to meet stringent pharmaceutical standards. Other methods require the pre-activation of substrates, such as converting nitro groups to amines in a separate step before cyclization, which increases waste generation and lowers the overall atom economy. These limitations collectively result in longer lead times and higher production costs, creating bottlenecks for the reliable pharmaceutical intermediate supplier trying to meet market demand.
The Novel Approach
In stark contrast, the novel approach detailed in patent CN112480015B utilizes a clever tandem reaction strategy that consolidates reduction, coupling, and cyclization into a single operational unit. By employing Molybdenum Hexacarbonyl (Mo(CO)6) as a solid carbon monoxide surrogate, the method eliminates the hazards associated with handling high-pressure CO gas, allowing the reaction to proceed in standard glassware or reactors. The use of nitro compounds as direct precursors is a game-changer, as these are among the cheapest and most abundant feedstocks in organic synthesis. The reaction tolerates a wide array of functional groups, including halogens and alkyl chains, ensuring that diverse analogues can be generated without protecting group manipulations. This versatility supports cost reduction in API manufacturing by minimizing purification steps and maximizing the yield of the desired heterocyclic core through a convergent synthetic design.
Mechanistic Insights into Pd-Catalyzed Carbonylation Cascade
The mechanistic pathway of this transformation is a sophisticated example of transition metal catalysis designed for maximum efficiency. The reaction initiates with the reduction of the nitro group on the aromatic substrate to an amine species, mediated by the Mo(CO)6 under thermal conditions. Simultaneously, the palladium catalyst, coordinated with the dppp ligand, facilitates the activation of the trifluoroethylimidoyl chloride. The newly formed amine then undergoes a base-promoted nucleophilic attack on the imidoyl chloride to form a trifluoroacetamidine intermediate in situ. Subsequently, the palladium center inserts into the carbon-iodine bond of the aromatic ring, followed by the insertion of carbon monoxide released from the molybdenum complex. This sequence generates an acyl-palladium species which undergoes intramolecular cyclization with the nitrogen atom, ultimately releasing the 2-trifluoromethyl quinazolinone product via reductive elimination. This intricate dance of organometallic steps ensures high regioselectivity and minimizes side reactions.
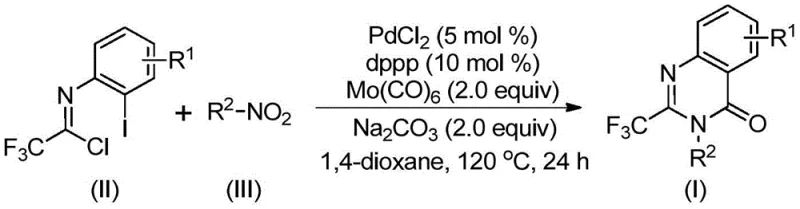
Understanding the impurity profile is crucial for R&D teams aiming to replicate this process at scale. The mechanism suggests that potential impurities could arise from incomplete reduction of the nitro group or hydrolysis of the imidoyl chloride. However, the use of sodium carbonate as a base helps to scavenge acidic byproducts and drive the equilibrium towards the cyclized product. The choice of 1,4-dioxane as the solvent is also critical, as it provides the necessary polarity to dissolve the inorganic base while stabilizing the organometallic intermediates. The patent data indicates that extending the reaction time beyond the optimal window does not significantly improve conversion but may lead to decomposition, highlighting the importance of precise process control. For those seeking high-purity pharmaceutical intermediates, this mechanistic understanding allows for the fine-tuning of reaction parameters to suppress trace impurities effectively.
How to Synthesize 2-Trifluoromethyl Quinazolinone Efficiently
Implementing this synthesis requires careful attention to the stoichiometry of the reagents and the quality of the catalyst system. The protocol specifies a molar ratio where the nitro compound is used in slight excess relative to the trifluoroethylimidoyl chloride to ensure complete consumption of the more valuable fluorinated building block. The catalyst loading is kept low, typically around 5 mol% for PdCl2, which is economically favorable for large-scale operations. The reaction is heated to 120°C, a temperature that balances the kinetic energy required for the carbonylation steps with the thermal stability of the reagents. Detailed standardized synthesis steps, including specific workup procedures like filtration and silica gel treatment, are outlined in the guide below to ensure reproducibility across different laboratory settings.
- Combine palladium chloride, dppp ligand, sodium carbonate, Mo(CO)6, trifluoroethylimidoyl chloride, and nitro compound in an organic solvent like dioxane.
- Heat the reaction mixture to 120°C and stir for 16 to 30 hours to allow the carbonylation cascade and cyclization to proceed.
- Upon completion, filter the mixture, mix with silica gel, and purify via column chromatography to isolate the target quinazolinone.
Commercial Advantages for Procurement and Supply Chain Teams
From a procurement perspective, this technology offers substantial advantages by decoupling production from volatile supply chains associated with high-pressure gases and exotic reagents. The reliance on commodity chemicals like nitrobenzenes and simple imidoyl chlorides means that sourcing is straightforward and less susceptible to geopolitical disruptions. The elimination of high-pressure equipment requirements significantly lowers the barrier to entry for contract manufacturing organizations (CMOs), allowing for more competitive bidding and reducing lead time for high-purity pharmaceutical intermediates. Furthermore, the one-pot nature of the reaction reduces solvent consumption and waste disposal costs, aligning with modern green chemistry initiatives that are increasingly mandated by regulatory bodies. This operational efficiency translates directly into a more resilient and cost-effective supply chain for downstream drug manufacturers.
- Cost Reduction in Manufacturing: The primary driver for cost savings lies in the replacement of expensive pre-activated substrates and high-pressure infrastructure with inexpensive nitro compounds and atmospheric pressure conditions. By avoiding the need for specialized autoclaves and reducing the number of isolation steps between reduction and cyclization, the overall processing time and labor costs are drastically simplified. Additionally, the use of a solid CO source like Mo(CO)6 removes the logistical complexities and safety costs associated with storing and transporting toxic carbon monoxide gas cylinders. These factors combine to create a manufacturing process that is inherently leaner and more economical, providing significant margin improvements for commercial production runs.
- Enhanced Supply Chain Reliability: The robustness of the starting materials ensures a stable supply chain, as nitro compounds and trifluoroethylimidoyl chlorides are widely produced bulk chemicals with multiple global suppliers. This diversity in sourcing options mitigates the risk of single-source dependency, which is a critical consideration for supply chain heads managing long-term projects. The tolerance of the reaction to various functional groups also means that supply chain disruptions for specific substituted anilines can be bypassed by switching to the corresponding nitro precursors, which are often more stable and easier to store. This flexibility ensures continuous production capability even when specific fine chemical feedstocks face temporary shortages.
- Scalability and Environmental Compliance: The process is designed with scalability in mind, having been demonstrated to work efficiently from milligram to gram scales with consistent yields. The absence of high-pressure gas and the use of standard organic solvents simplify the engineering controls required for scale-up to multi-kilogram or tonne batches. From an environmental standpoint, the atom economy is improved by the direct use of nitro groups, which avoids the generation of stoichiometric amounts of reducing agent waste typical in separate reduction steps. The simplified workup involving filtration and chromatography reduces the volume of aqueous waste streams, facilitating easier compliance with environmental regulations and lowering the cost of waste treatment facilities.
Frequently Asked Questions (FAQ)
The following questions address common technical inquiries regarding the implementation of this synthesis method, derived directly from the experimental data and scope defined in the patent documentation. These insights are intended to assist technical teams in evaluating the feasibility of adopting this route for their specific project needs. Understanding the nuances of catalyst loading, substrate compatibility, and reaction conditions is essential for successful technology transfer from the laboratory to pilot plant operations.
Q: What is the carbon monoxide source in this synthesis?
A: The method utilizes Molybdenum Hexacarbonyl (Mo(CO)6) as a solid, easy-to-handle carbon monoxide substitute, eliminating the need for high-pressure gas cylinders.
Q: Does this method support diverse substrate scopes?
A: Yes, the protocol demonstrates excellent compatibility with various substituents including halogens, alkyl groups, and trifluoromethyl groups on both the aromatic ring and the nitrogen substituent.
Q: What catalyst system is employed for this transformation?
A: The reaction employs a Palladium(II) Chloride (PdCl2) catalyst coordinated with 1,3-bis(diphenylphosphino)propane (dppp) as the ligand.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 2-Trifluoromethyl Quinazolinone Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical role that advanced synthetic methodologies play in accelerating drug discovery and development. Our team of expert process chemists has extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the transition from benchtop to manufacturing is seamless and efficient. We are committed to delivering products that meet stringent purity specifications, supported by our rigorous QC labs equipped with state-of-the-art analytical instrumentation. Whether you require custom synthesis of novel quinazolinone analogues or reliable supply of established intermediates, our infrastructure is designed to support your most demanding projects with speed and precision.
We invite you to contact our technical procurement team to discuss how this innovative one-pot synthesis can be integrated into your supply chain. By requesting a Customized Cost-Saving Analysis, you can gain deeper insights into the economic benefits of switching to this methodology for your specific targets. We encourage potential partners to reach out for specific COA data and route feasibility assessments, allowing us to demonstrate our capability to deliver high-quality 2-trifluoromethyl quinazolinones that meet your exact requirements.