Advanced Synthesis of Montelukast Sodium Intermediate: A Technical Breakthrough for Scalable Pharma Manufacturing
Advanced Synthesis of Montelukast Sodium Intermediate: A Technical Breakthrough for Scalable Pharma Manufacturing
The pharmaceutical industry constantly seeks more efficient pathways for producing critical asthma medications, and the synthesis of Montelukast Sodium intermediates remains a focal point for process optimization. Patent CN115710221A introduces a transformative four-step methodology that addresses the longstanding challenges of low yield and complex purification associated with traditional routes. This technical insight report analyzes the novel condensation and coupling strategies that enable the production of high-purity 2-[3-(S)-[3-(2-(7-chloro-2-quinolyl) vinyl) phenyl]-3-hydroxypropyl] phenyl-2-propanol. By leveraging commercially available raw materials and implementing a closed-loop iodine recovery system, this method offers a compelling value proposition for reliable montelukast sodium intermediate supplier networks aiming to enhance supply chain resilience. The following analysis details the mechanistic advantages and commercial implications of adopting this streamlined synthetic approach.
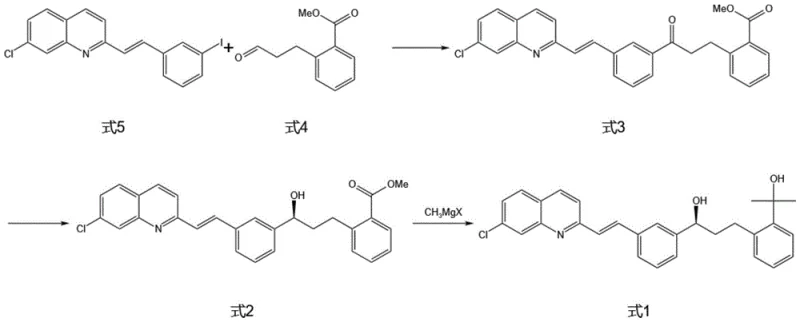
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the manufacturing of Montelukast intermediates has been plagued by inefficient multi-step sequences that erode overall process economics. Prior art, such as the methods disclosed in CN101638381A, relies on the condensation of 3-cyanobenzaldehyde with 7-chloroquinaldine to form a nitrile intermediate, which subsequently undergoes further transformation. This pathway is inherently flawed due to the excessive number of reaction steps, which cumulatively degrade the overall yield and increase the accumulation of difficult-to-remove impurities. Furthermore, alternative routes described in WO2008035086 utilize ester precursors that exhibit poor conversion rates during the critical transformation to tertiary alcohol groups. These conventional methodologies often necessitate the use of hazardous reagents and result in significant material loss, creating a bottleneck for cost reduction in pharmaceutical intermediates manufacturing. The reliance on non-recyclable stoichiometric reagents and the generation of toxic waste streams further complicate the environmental compliance profile of these legacy processes.
The Novel Approach
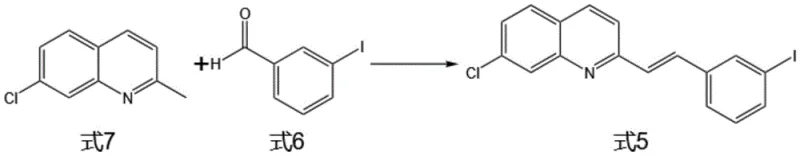
In stark contrast, the methodology outlined in CN115710221A presents a concise and robust alternative that drastically simplifies the production landscape. The new route initiates with a direct condensation of 7-chloroquinaldine and 3-iodobenzaldehyde, bypassing the need for complex nitrile handling. A key innovation lies in the subsequent palladium-catalyzed coupling with a keto-ester derivative, which efficiently constructs the carbon skeleton in fewer operations. This approach not only shortens the production timeline but also significantly enhances the stereochemical integrity of the final product. By utilizing selective reducing agents and optimizing reaction conditions, the novel method achieves superior conversion rates without the extensive purification burdens seen in older patents. This streamlined workflow directly supports the commercial scale-up of complex pharmaceutical intermediates by reducing the operational footprint and minimizing the risk of batch failures due to cumulative yield losses.
Mechanistic Insights into Palladium-Catalyzed Coupling and Chiral Reduction
The core of this synthetic breakthrough resides in the precise orchestration of catalytic cycles and stereoselective transformations. The second step involves a sophisticated palladium-catalyzed reaction between the vinyl quinoline intermediate (Formula 5) and the keto-ester (Formula 4). The use of palladium acetate in conjunction with a phase transfer catalyst like benzyltriethylammonium bromide facilitates a smooth cross-coupling under mild thermal conditions. This catalytic system is crucial for maintaining the integrity of the sensitive vinyl group while ensuring high regioselectivity. Following this, the establishment of the chiral center is achieved through a highly specific reduction using (-)-diisopinocampheylchloroborane. This chiral borane reagent delivers hydride to the ketone face with exceptional fidelity, ensuring the formation of the desired (S)-enantiomer with minimal racemization. Such mechanistic precision is vital for meeting the stringent regulatory requirements for high-purity montelukast sodium intermediate specifications, as even trace amounts of the wrong enantiomer can compromise the therapeutic efficacy of the final drug product.
Impurity control is further enhanced by the strategic selection of solvents and reaction parameters throughout the sequence. For instance, the initial condensation in acetic anhydride promotes high solubility and reaction kinetics, minimizing the formation of oligomeric by-products. The subsequent workup procedures, including specific washing protocols with sodium sulfite and carbonate solutions, are designed to effectively remove residual metal catalysts and acidic impurities. The final Grignard addition is mediated by an organotitanium reagent generated in situ, which moderates the reactivity of the methyl magnesium halide to prevent over-addition or attack on other sensitive functional groups. This layered approach to impurity management ensures that the final crystalline product exhibits a purity profile that exceeds 99%, thereby reducing lead time for high-purity pharmaceutical intermediates by eliminating the need for repetitive recrystallization or chromatographic purification steps.
How to Synthesize Montelukast Sodium Intermediate Efficiently
The implementation of this synthesis requires strict adherence to the optimized conditions detailed in the patent to maximize yield and safety. The process begins with the preparation of the vinyl quinoline backbone, followed by the critical coupling and reduction steps that define the molecule's chirality. Operators must maintain precise temperature controls, particularly during the exothermic Grignard addition and the low-temperature chiral reduction, to ensure reproducibility. The following guide outlines the standardized operational framework derived from the patent examples, providing a clear roadmap for technical teams to replicate these results in a pilot or production setting.
- Condense 7-chloroquinaldine (Formula 7) with 3-iodobenzaldehyde (Formula 6) in acetic anhydride to form the vinyl quinoline intermediate (Formula 5).
- Perform a palladium-catalyzed coupling between Formula 5 and methyl 2-(3-oxopropyl)benzoate (Formula 4) using a phase transfer catalyst to generate the ketone precursor (Formula 3).
- Execute a highly selective asymmetric reduction of Formula 3 using (-)-diisopinocampheylchloroborane to establish the critical chiral center, yielding Formula 2.
- React Formula 2 with methyl magnesium halide in the presence of an organotitanium reagent to finalize the tertiary alcohol structure of the target intermediate (Formula 1).
Commercial Advantages for Procurement and Supply Chain Teams
From a procurement and supply chain perspective, this novel synthesis route offers substantial strategic benefits that extend beyond mere technical feasibility. The primary advantage lies in the sourcing strategy; all key starting materials, including 7-chloroquinaldine and 3-iodobenzaldehyde, are commercially available commodities. This eliminates the need for costly and time-consuming in-house synthesis of precursors, thereby stabilizing the supply chain against raw material volatility. Moreover, the process incorporates a clever iodine recovery loop, where iodine by-products are reclaimed and converted back into 3-iodobenzaldehyde. This circular economy approach significantly lowers the net consumption of expensive halogenated reagents, driving down the variable cost per kilogram of the final intermediate without compromising quality.
- Cost Reduction in Manufacturing: The elimination of toxic solvents like acetonitrile and the reduction in total reaction steps directly translate to lower operational expenditures. By avoiding the complex multi-step sequences of prior art, manufacturers save on energy consumption, solvent disposal, and labor hours. The high conversion rates observed in the patent examples mean that less raw material is wasted as unreacted feedstock or side products, leading to a more efficient utilization of capital. Furthermore, the ability to recover and reuse iodine species creates a closed-loop system that insulates production costs from fluctuations in the global iodine market, ensuring long-term economic stability for the manufacturing process.
- Enhanced Supply Chain Reliability: Dependence on custom-synthesized or hard-to-source intermediates is a major risk factor in pharmaceutical supply chains. This new method mitigates that risk by relying on robust, off-the-shelf chemicals that have established global supply networks. The simplified workflow also reduces the likelihood of bottlenecks at any single stage of production, as each step is designed for high throughput and reliability. This resilience is critical for maintaining continuous supply to downstream API manufacturers, preventing stockouts that could disrupt the production of life-saving asthma medications. The robustness of the chemistry ensures that scale-up from pilot to commercial volumes can be achieved with minimal process re-engineering.
- Scalability and Environmental Compliance: Modern manufacturing demands adherence to strict environmental, health, and safety (EHS) standards. This synthesis route aligns perfectly with green chemistry principles by replacing hazardous reagents with safer alternatives and minimizing waste generation. The absence of highly toxic cyanide intermediates, which were prevalent in older methods, significantly reduces the regulatory burden and the cost of hazardous waste treatment. The process is inherently scalable, as demonstrated by the use of common industrial solvents like toluene and ethyl acetate, which are easy to handle and recycle in large-scale reactors. This environmental compatibility not only future-proofs the manufacturing site against tightening regulations but also enhances the corporate sustainability profile of the supply chain.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis method. These answers are derived directly from the experimental data and beneficial effects reported in the patent documentation, providing clarity on the practical aspects of adoption. Understanding these nuances is essential for R&D and procurement teams evaluating the feasibility of integrating this technology into their existing manufacturing portfolios.
Q: How does this new synthesis route improve product purity compared to prior art?
A: The novel route utilizes highly stereoselective reagents like (-)-diisopinocampheylchloroborane for the reduction step, minimizing racemic impurities. Additionally, the avoidance of harsh cyanide-based intermediates found in older methods significantly reduces side-reaction by-products, resulting in final purities exceeding 99%.
Q: What are the key cost-saving drivers in this manufacturing process?
A: Cost efficiency is driven by the use of commercially available starting materials that require no pre-synthesis. Furthermore, the process incorporates an iodine recovery loop from the reaction waste, allowing for the regeneration of 3-iodobenzaldehyde, which drastically lowers raw material consumption and waste disposal costs.
Q: Is this process suitable for large-scale commercial production?
A: Yes, the process is designed for scalability. It eliminates the use of highly toxic solvents like acetonitrile and replaces them with safer alternatives like toluene and ethyl acetate. The reaction conditions are moderate, and the high conversion rates reduce the burden on downstream purification, making it ideal for metric-ton scale manufacturing.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Montelukast Sodium Intermediate Supplier
The technical advancements detailed in patent CN115710221A represent a significant leap forward in the production of Montelukast Sodium intermediates, offering a pathway to higher purity and lower costs. At NINGBO INNO PHARMCHEM, we possess the extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production required to bring this innovative chemistry to the global market. Our state-of-the-art facilities are equipped with rigorous QC labs capable of verifying stringent purity specifications, ensuring that every batch meets the exacting standards required by top-tier pharmaceutical companies. We are committed to leveraging this advanced synthesis to provide a consistent, high-quality supply of this critical intermediate.
We invite potential partners to engage with our technical procurement team to discuss how this optimized route can benefit your specific supply chain needs. By requesting a Customized Cost-Saving Analysis, you can gain deeper insights into the economic advantages of switching to this newer methodology. We encourage you to contact us today to obtain specific COA data and route feasibility assessments tailored to your project requirements, ensuring a seamless transition to a more efficient and reliable sourcing strategy.