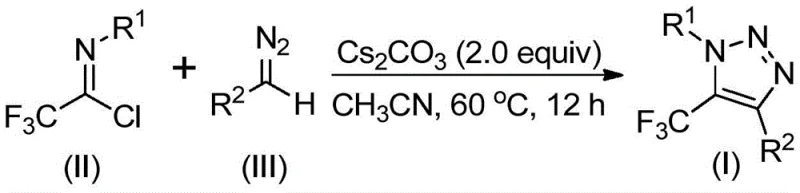
Scalable Metal-Free Synthesis of 5-Trifluoromethyl-1,2,3-Triazoles for Advanced Drug Discovery
The pharmaceutical and agrochemical industries are constantly seeking robust methodologies to construct nitrogen-containing heterocycles, particularly those incorporating fluorine motifs which enhance metabolic stability and bioavailability. Patent CN113121462A introduces a groundbreaking preparation method for 5-trifluoromethyl substituted 1,2,3-triazole compounds, addressing critical bottlenecks in current synthetic workflows. This innovation leverages a base-promoted cyclization strategy that bypasses the need for transition metal catalysts and hazardous azide reagents, marking a significant shift towards greener and safer chemical manufacturing. By utilizing trifluoroethylimidoyl chloride and diazo compounds as key building blocks, the process achieves high reaction efficiency under mild thermal conditions. For R&D directors and procurement specialists, this represents a pivotal opportunity to streamline the supply chain for high-purity pharmaceutical intermediates while mitigating the risks associated with heavy metal contamination and explosive precursors.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the construction of trifluoromethyl-substituted triazoles has relied heavily on copper-catalyzed [3+2] cycloaddition reactions between alkynes and organic azides, followed by trifluoromethylation steps. These traditional pathways present severe drawbacks, primarily centering on the use of toxic and potentially explosive organic azides which pose significant safety hazards during storage and handling on a large scale. Furthermore, the reliance on copper catalysts necessitates rigorous downstream purification processes to remove residual heavy metals to meet stringent pharmaceutical regulatory standards, thereby increasing both production time and costs. Alternative methods involving 1,3-dipolar cycloaddition of azides with trifluoromethyl ketones also suffer from similar safety profiles and often require harsh reaction conditions that limit functional group tolerance. These legacy techniques create substantial barriers for reliable pharmaceutical intermediate suppliers aiming to deliver consistent quality without compromising on safety or environmental compliance.
The Novel Approach
In stark contrast, the methodology disclosed in CN113121462A utilizes a metal-free, base-promoted strategy that fundamentally alters the risk profile of triazole synthesis. By reacting trifluoroethylimidoyl chloride with diazo compounds in the presence of cesium carbonate, the process eliminates the need for explosive azides and toxic transition metals entirely. This novel approach operates under remarkably mild conditions, typically between 50°C and 70°C, which preserves sensitive functional groups and reduces energy consumption compared to high-temperature alternatives. The reaction proceeds through an intermolecular nucleophilic addition-elimination mechanism followed by intramolecular cyclization, offering a direct and atom-economical route to the target scaffold. This shift not only simplifies the operational workflow but also drastically reduces the complexity of post-reaction workup, making it an ideal candidate for cost reduction in pharmaceutical intermediate manufacturing.
Mechanistic Insights into Base-Promoted Cyclization
The core of this synthetic breakthrough lies in the unique reactivity of the diazo compound towards the electrophilic carbon of the trifluoroethylimidoyl chloride. Under the promotion of a mild inorganic base like cesium carbonate, the diazo species acts as a nucleophile, attacking the imidoyl chloride to form a transient intermediate. This initial step is crucial as it establishes the carbon-carbon bond necessary for ring closure without requiring external activation by expensive ligands or metals. The subsequent intramolecular 5-endo-dig cyclization is facilitated by the electron-withdrawing nature of the trifluoromethyl group, which stabilizes the transition state and drives the formation of the five-membered triazole ring. This mechanistic pathway ensures high regioselectivity, predominantly yielding the 1,4-substituted isomer which is often the desired pharmacophore in medicinal chemistry applications. Understanding this mechanism allows chemists to fine-tune reaction parameters for optimal yield and purity.
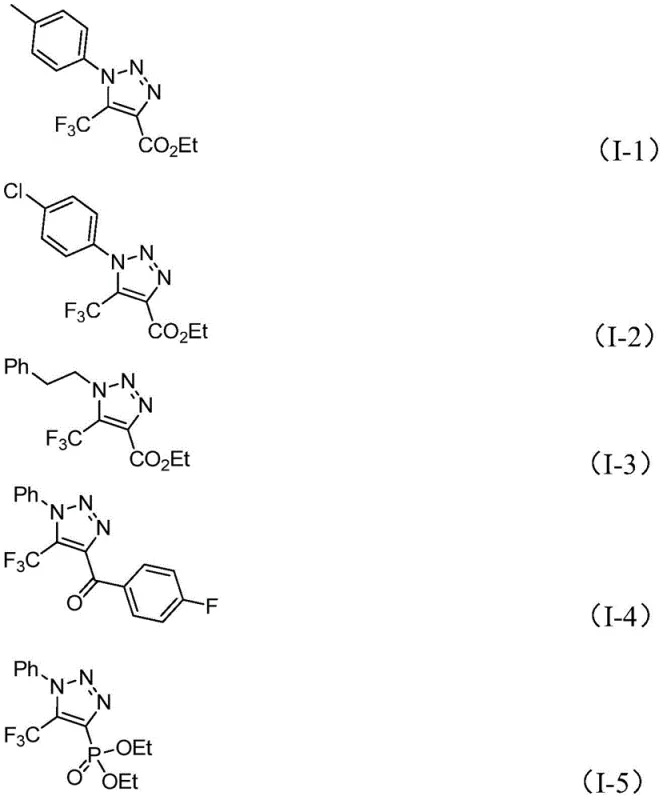
Furthermore, the choice of solvent and base plays a pivotal role in controlling the impurity profile of the final product. The patent highlights acetonitrile as the preferred solvent due to its ability to effectively dissolve both organic substrates and the inorganic base, ensuring homogeneous reaction conditions that minimize side reactions. The use of molecular sieves in the reaction mixture serves to scavenge trace moisture, which could otherwise hydrolyze the sensitive imidoyl chloride or diazo reagents, leading to decreased yields and complex impurity spectra. By strictly controlling these variables, the process achieves a high degree of reproducibility and scalability. The broad substrate scope demonstrated in the patent, accommodating various aryl and aroyl groups, confirms the robustness of this mechanistic framework against steric and electronic variations, providing a versatile platform for synthesizing diverse libraries of bioactive molecules.
How to Synthesize 5-Trifluoromethyl-1,2,3-Triazole Efficiently
To implement this synthesis in a laboratory or pilot plant setting, precise adherence to the molar ratios and reaction conditions specified in the patent is essential for maximizing yield. The standard protocol involves combining the trifluoroethylimidoyl chloride and the specific diazo compound in acetonitrile with cesium carbonate acting as the promoter. Detailed standardized synthesis steps see the guide below.
- Mix cesium carbonate, molecular sieves, trifluoroethylimidoyl chloride, and diazo compound in an organic solvent such as acetonitrile.
- Heat the reaction mixture to a temperature between 50°C and 70°C and maintain stirring for 8 to 16 hours to ensure complete conversion.
- Filter the reaction mixture, mix with silica gel, and purify via column chromatography to isolate the final 5-trifluoromethyl substituted 1,2,3-triazole product.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this metal-free synthesis route offers transformative benefits that extend beyond simple yield improvements. The elimination of copper catalysts removes the necessity for expensive and time-consuming heavy metal scavenging steps, which are often a bottleneck in the production of GMP-grade intermediates. This simplification of the downstream processing directly translates to reduced operational expenditures and shorter batch cycle times. Additionally, the starting materials, including cesium carbonate and various diazo precursors, are commercially available and relatively inexpensive, ensuring a stable and cost-effective supply chain. The mild reaction temperatures further contribute to energy savings and reduce the thermal load on reactor systems, enhancing overall process safety and sustainability.
- Cost Reduction in Manufacturing: The removal of transition metal catalysts significantly lowers the cost of goods sold by eliminating the need for specialized ligands and extensive purification protocols required to meet residual metal limits. The use of cheap inorganic bases like cesium carbonate instead of expensive organometallic reagents further drives down raw material costs. Moreover, the high atom economy of the reaction minimizes waste generation, reducing disposal costs and environmental fees associated with hazardous chemical byproducts. These factors collectively create a leaner manufacturing process that is highly competitive in the global market for fine chemicals.
- Enhanced Supply Chain Reliability: By relying on stable and readily available starting materials rather than hazardous azides or sensitive organometallics, the risk of supply disruptions due to safety regulations or transportation restrictions is substantially mitigated. The robustness of the reaction conditions allows for flexible scheduling and easier scale-up from gram to kilogram quantities without significant re-optimization. This reliability ensures consistent delivery timelines for downstream customers, fostering stronger long-term partnerships and reducing the inventory buffer stocks that buyers typically hold to guard against supply volatility.
- Scalability and Environmental Compliance: The process is inherently scalable, having been demonstrated to work efficiently at gram levels with the potential for ton-scale production, making it suitable for commercial manufacturing of complex pharmaceutical intermediates. The absence of heavy metals and explosive reagents simplifies waste treatment and aligns with increasingly stringent environmental regulations regarding green chemistry principles. This compliance advantage future-proofs the supply chain against evolving regulatory landscapes, ensuring uninterrupted production capabilities and maintaining the social license to operate in highly regulated jurisdictions.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this patented synthesis method. These answers are derived directly from the experimental data and beneficial effects described in the patent documentation to provide clarity for potential partners.
Q: What are the primary safety advantages of this synthesis method compared to traditional routes?
A: Unlike conventional methods that rely on toxic and explosive organic azides or copper catalysts requiring removal, this patent utilizes stable diazo compounds and imidoyl chlorides under metal-free conditions, significantly enhancing operational safety and reducing hazardous waste.
Q: Can this process be scaled for industrial production of pharmaceutical intermediates?
A: Yes, the patent explicitly states that the method can be expanded to gram-level reactions and potentially industrial scale due to its mild conditions (50-70°C), simple operation, and the use of commercially available, inexpensive starting materials like cesium carbonate.
Q: What is the substrate scope for the R1 and R2 groups in this triazole synthesis?
A: The method demonstrates broad tolerance, accommodating various substituents including alkyl, substituted or unsubstituted aryl groups for R1, and aroyl, phospholipid, alkoxycarbonyl, or trifluoromethyl groups for R2, allowing for diverse molecular scaffolding.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 5-Trifluoromethyl-1,2,3-Triazole Supplier
At NINGBO INNO PHARMCHEM, we recognize the strategic value of adopting innovative synthetic routes like the one described in CN113121462A to enhance our portfolio of high-purity pharmaceutical intermediates. As a dedicated CDMO partner, we possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your project transitions smoothly from benchtop discovery to full-scale manufacturing. Our facilities are equipped with rigorous QC labs and advanced analytical instrumentation to guarantee stringent purity specifications, particularly critical when removing trace impurities in metal-free processes. We are committed to delivering consistent quality and supply continuity for complex heterocyclic building blocks.
We invite you to collaborate with us to leverage this efficient synthesis technology for your next drug development program. Our technical team is ready to provide a Customized Cost-Saving Analysis tailored to your specific volume requirements and purity needs. Please contact our technical procurement team today to request specific COA data for our triazole derivatives and discuss route feasibility assessments for your custom synthesis projects. Together, we can accelerate your timeline to market while optimizing your manufacturing costs.