Scalable Metal-Free Synthesis of 3,4,5-Trisubstituted 1,2,4-Triazoles for Pharmaceutical Applications
Scalable Metal-Free Synthesis of 3,4,5-Trisubstituted 1,2,4-Triazoles for Pharmaceutical Applications
The pharmaceutical industry continuously seeks robust synthetic methodologies for constructing nitrogen-rich heterocyclic scaffolds, particularly the 1,2,4-triazole motif, which serves as a critical pharmacophore in numerous high-value therapeutic agents. As illustrated in the structural diversity of bioactive molecules such as Maraviroc, Sitagliptin, and Deferasirox shown below, the incorporation of a trifluoromethyl group into these heterocycles can drastically enhance metabolic stability, lipophilicity, and bioavailability. 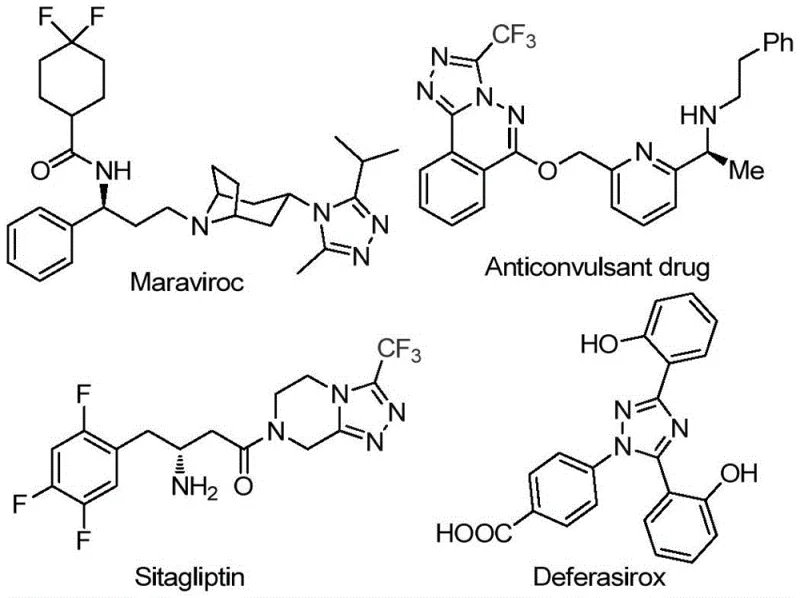 . Patent CN113105402B discloses a groundbreaking preparation method for 3,4,5-trisubstituted 1,2,4-triazole compounds that addresses long-standing challenges in heterocyclic chemistry. This innovation provides a streamlined, metal-free pathway that utilizes inexpensive aryl ethyl ketones and trifluoroethylimide hydrazides as starting materials. For R&D directors and procurement specialists, this technology represents a significant opportunity to optimize the supply chain for complex API intermediates by removing dependency on precious metal catalysts and simplifying purification protocols.
. Patent CN113105402B discloses a groundbreaking preparation method for 3,4,5-trisubstituted 1,2,4-triazole compounds that addresses long-standing challenges in heterocyclic chemistry. This innovation provides a streamlined, metal-free pathway that utilizes inexpensive aryl ethyl ketones and trifluoroethylimide hydrazides as starting materials. For R&D directors and procurement specialists, this technology represents a significant opportunity to optimize the supply chain for complex API intermediates by removing dependency on precious metal catalysts and simplifying purification protocols.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the construction of polysubstituted 1,2,4-triazole rings, especially those bearing both acyl and trifluoromethyl groups, has relied heavily on transition metal catalysis or harsh reaction conditions that are ill-suited for large-scale manufacturing. Traditional routes often necessitate the use of expensive copper or palladium catalysts, which introduce significant cost burdens and create severe regulatory hurdles regarding heavy metal residues in final drug substances. Furthermore, many existing protocols require stringent anhydrous and oxygen-free environments, demanding specialized equipment and increasing the operational expenditure for chemical production facilities. The limited functional group tolerance in some classical methods also restricts the structural diversity accessible to medicinal chemists, forcing them to adopt longer, less efficient linear syntheses to access specific substitution patterns on the triazole core.
The Novel Approach
In stark contrast, the methodology outlined in patent CN113105402B leverages a non-metallic iodine-promoted strategy that operates under remarkably mild and operationally simple conditions. By utilizing dimethyl sulfoxide (DMSO) as both solvent and reactant participant, this novel approach facilitates an iodination and Kornblum oxidation sequence in situ, generating the necessary reactive intermediates without external oxidants. The reaction does not require inert atmospheres or rigorously dried solvents, making it highly attractive for cost reduction in pharmaceutical intermediate manufacturing. This shift from precious metal catalysis to abundant halogen promotion not only lowers the raw material costs but also drastically simplifies the post-reaction workup, allowing for easier isolation of high-purity products suitable for downstream coupling reactions in API synthesis.
Mechanistic Insights into Iodine-Promoted Tandem Cyclization
The mechanistic pathway of this transformation is a sophisticated tandem sequence that begins with the activation of the aryl ethyl ketone substrate. Initially, elemental iodine interacts with the ketone in the presence of DMSO to effect a Kornblum oxidation, converting the methyl ketone into a reactive alpha-dicarbonyl or alpha-iodo ketone species. This activated electrophile then undergoes a condensation reaction with the trifluoroethylimide hydrazide to form a hydrazone intermediate. Subsequently, under the continued influence of iodine and the basic environment provided by pyridine and sodium dihydrogen phosphate, an intramolecular cyclization occurs. This cascade effectively constructs the 1,2,4-triazole ring while simultaneously installing the trifluoromethyl group at the 3-position and the acyl group at the 5-position, as depicted in the general reaction scheme below. 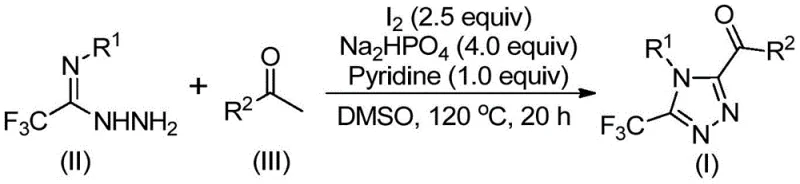 .
.
From an impurity control perspective, this mechanism offers distinct advantages due to the high chemoselectivity of the iodine-mediated oxidation. The use of sodium dihydrogen phosphate acts as a buffer to maintain optimal pH levels, preventing the degradation of sensitive functional groups on the aromatic rings of the substrates. The reaction tolerates a wide range of substituents, including electron-donating groups like methoxy and methyl, as well as electron-withdrawing groups such as chloro and trifluoromethyl, without significant formation of side products. This broad substrate scope ensures that the impurity profile remains manageable, reducing the burden on analytical teams during method validation and ensuring consistent quality for commercial scale-up of complex pharmaceutical intermediates.
How to Synthesize 3,4,5-Trisubstituted 1,2,4-Triazole Compounds Efficiently
The practical execution of this synthesis involves a two-stage heating protocol within a standard reaction vessel, eliminating the need for specialized high-pressure equipment. The process begins by combining the aryl ethyl ketone and a stoichiometric amount of iodine in DMSO, followed by an initial heating phase to generate the oxidized intermediate. Once this conversion is underway, the trifluoroethylimide hydrazide, along with the base system and additional iodine, is introduced to drive the cyclization to completion. The detailed standardized synthetic steps, including precise molar ratios and temperature gradients optimized for maximum yield, are provided in the guide below.
- Oxidize aryl ethyl ketone with iodine in DMSO at 90-110°C for 4-6 hours to form the reactive dicarbonyl intermediate.
- Add trifluoroethylimide hydrazide, sodium dihydrogen phosphate, pyridine, and additional iodine to the mixture.
- Heat the reaction to 110-130°C for 12-20 hours to complete the cyclization, followed by filtration and column chromatography.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this iodine-promoted synthesis offers transformative benefits regarding cost structure and logistical reliability. The elimination of transition metal catalysts removes a major cost driver associated with both the purchase of noble metals and the subsequent scavenging processes required to meet regulatory limits. Furthermore, the reliance on commodity chemicals like acetophenones and elemental iodine ensures a stable and resilient supply chain, insulating production schedules from the volatility often seen in the market for specialized organometallic reagents. This robustness allows for more accurate forecasting and inventory management, crucial for maintaining continuity in API production lines.
- Cost Reduction in Manufacturing: The economic impact of switching to this metal-free protocol is substantial, primarily driven by the removal of expensive catalyst systems and the simplification of purification workflows. Without the need for silica-supported metal scavengers or complex extraction procedures to remove heavy metals, the overall processing time and consumable costs are significantly reduced. Additionally, the high atom economy of using simple ketones and hydrazides means that raw material expenditures are minimized, directly improving the gross margin for the final intermediate product.
- Enhanced Supply Chain Reliability: Sourcing strategies are greatly improved as the key starting materials, such as substituted acetophenones and trifluoroacetic acid derivatives, are globally available commodity chemicals with multiple qualified suppliers. This diversification of the supply base reduces the risk of single-source bottlenecks that can halt production. The operational simplicity of the reaction, which tolerates ambient moisture and oxygen, further enhances reliability by reducing the likelihood of batch failures due to environmental excursions, ensuring consistent delivery timelines to downstream customers.
- Scalability and Environmental Compliance: The process is inherently scalable, having been demonstrated effectively at the gram level with clear pathways to kilogram and tonne-scale production. The use of DMSO, a high-boiling polar aprotic solvent, facilitates heat transfer and mixing in large reactors, while the absence of toxic heavy metals simplifies waste stream treatment and disposal. This alignment with green chemistry principles not only reduces environmental compliance costs but also supports corporate sustainability goals, making the manufacturing process more attractive to environmentally conscious partners.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis technology. These insights are derived directly from the experimental data and beneficial effects reported in the patent documentation, providing clarity on reaction scope, operational parameters, and scalability potential for industrial partners evaluating this route for their own manufacturing portfolios.
Q: What are the primary advantages of this iodine-promoted method over traditional metal-catalyzed routes?
A: This method eliminates the need for expensive and toxic transition metal catalysts, significantly simplifying downstream purification and reducing heavy metal residue risks in pharmaceutical intermediates.
Q: Does this synthesis require strict anhydrous or oxygen-free conditions?
A: No, one of the key operational benefits described in patent CN113105402B is that the reaction proceeds efficiently without the need for rigorous anhydrous or inert atmosphere conditions, lowering operational complexity.
Q: Is this process suitable for large-scale industrial production?
A: Yes, the patent explicitly demonstrates scalability from gram-level laboratory synthesis to potential industrial application, utilizing cheap and readily available starting materials like aryl ethyl ketones.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 3,4,5-Trisubstituted 1,2,4-Triazole Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical role that efficient heterocyclic synthesis plays in accelerating drug development timelines. Our technical team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the transition from laboratory discovery to industrial supply is seamless. We adhere to stringent purity specifications and utilize rigorous QC labs to guarantee that every batch of 3,4,5-trisubstituted 1,2,4-triazole intermediate meets the exacting standards required for pharmaceutical applications, providing our partners with absolute confidence in material quality.
We invite you to engage with our technical procurement team to discuss how this innovative metal-free technology can be integrated into your specific project requirements. By requesting a Customized Cost-Saving Analysis, you can gain a deeper understanding of the economic benefits tailored to your volume needs. We encourage you to contact us today to obtain specific COA data and route feasibility assessments, allowing us to demonstrate our commitment to being your trusted partner in advanced chemical manufacturing.