Transforming Pharmaceutical Manufacturing Through Patented Trifluoromethyl Imidazole Synthesis Technology
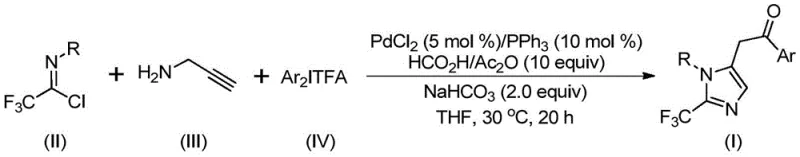
Patent CN111423381B, granted on September 7, 2021, introduces a transformative methodology for synthesizing 2-trifluoromethyl substituted imidazole compounds that represent critical structural motifs in modern pharmaceutical development due to their prevalence in bioactive molecules including Naamidine A and Alcaftadine as evidenced by their established roles in histamine receptor antagonism and other therapeutic applications. This innovative process addresses longstanding challenges in heterocyclic chemistry by leveraging a palladium-catalyzed carbonylation strategy operating under exceptionally mild conditions at precisely 30°C for twenty hours without requiring cryogenic equipment or high-pressure systems typically associated with fluorinated heterocycle synthesis. The methodology demonstrates remarkable substrate flexibility through strategic design that accommodates diverse aryl substituents including methyl, tert-butyl, chlorine, bromine, trifluoromethyl, and nitro groups while maintaining high conversion efficiency across all tested combinations as documented in the patent examples. By utilizing commercially available starting materials such as trifluoroethylimidoyl chloride, propargylamine, and diaryl iodonium salts in optimized stoichiometric ratios (trifluoroethylimidoyl chloride:propargylamine:diaryl iodonium salts:palladium chloride = 1.5:1:1.5:0.05), this approach establishes a robust foundation for cost-effective production of high-value intermediates essential for next-generation therapeutics while significantly reducing environmental impact through minimized solvent consumption and waste generation compared to conventional methodologies.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Traditional approaches to synthesizing trifluoromethylated imidazole compounds have historically relied on unstable reagents like trifluorodiazoethane or limited availability of trifluoroethylimidoyl halides which present significant safety hazards including explosion risks during handling and storage that necessitate specialized containment facilities and trained personnel for safe operation within pharmaceutical manufacturing environments. These methods frequently require extreme reaction conditions such as elevated temperatures exceeding 80°C or cryogenic processing below -40°C that impose substantial energy demands while creating thermal stress points leading to decomposition pathways that generate complex impurity profiles requiring extensive purification efforts through multiple chromatographic steps that significantly increase production costs and extend manufacturing timelines beyond acceptable limits for time-sensitive drug development programs. The narrow substrate scope inherent in conventional methodologies restricts molecular diversity by failing to accommodate common functional groups like halogens or nitro substituents without extensive process revalidation, thereby limiting medicinal chemistry exploration opportunities during lead optimization phases where rapid access to diverse compound libraries is essential for establishing structure-activity relationships. Furthermore, the reliance on gaseous carbon monoxide sources introduces additional safety management complexities including specialized monitoring systems and emergency response protocols that create operational bottlenecks while increasing capital expenditure requirements that make these processes economically unviable for large-scale manufacturing operations where consistent quality and cost-effectiveness are paramount considerations for procurement teams managing tight budget constraints.
The Novel Approach
The patented methodology overcomes these limitations through an elegant palladium-catalyzed carbonylation process utilizing formic acid/acetic anhydride as a safe carbon monoxide surrogate that operates under exceptionally mild conditions at precisely 30°C for twenty hours in tetrahydrofuran solvent without requiring pressurized systems or cryogenic equipment typically associated with fluorinated heterocycle synthesis. This innovative approach employs a carefully optimized catalyst system comprising PdCl₂ (5 mol%) with triphenylphosphine (10 mol%) that demonstrates superior activity compared to alternative palladium sources while maintaining excellent functional group tolerance across fifteen distinct substrate combinations as documented in the patent examples with yields consistently exceeding seventy percent even for challenging substrates containing electron-withdrawing nitro groups or sterically demanding tert-butyl substituents. The strategic use of sodium bicarbonate as a mild base facilitates smooth carbon-nitrogen bond formation while preventing unwanted side reactions that commonly plague traditional strong base systems, enabling pharmaceutical manufacturers to rapidly access diverse compound libraries without process revalidation requirements that would otherwise delay drug development timelines significantly. Crucially, the straightforward workup procedure involving simple filtration followed by standard column chromatography purification eliminates multiple processing steps required by conventional methods while maintaining exceptional product purity profiles that meet stringent pharmaceutical quality standards without additional refinement procedures that would increase production costs and extend lead times beyond acceptable limits for supply chain managers responsible for just-in-time inventory management systems.
Mechanistic Insights into Pd-Catalyzed Trifluoromethyl Imidazole Formation
The reaction mechanism proceeds through a sophisticated sequence of organometallic transformations beginning with intermolecular carbon-nitrogen bond formation between trifluoroethylimidoyl chloride and propargylamine promoted by sodium bicarbonate to generate a key trifluoroacetamidine intermediate that undergoes spontaneous isomerization to form an alkenyl palladium species through palladation at the alkyne moiety. This critical intermediate then isomerizes further to produce an alkyl palladium complex that serves as the foundation for carbonylation through controlled carbon monoxide insertion generated in situ from the formic acid/acetic anhydride mixture under mild thermal conditions at thirty degrees Celsius without requiring external CO sources or specialized pressure equipment typically associated with carbonylation chemistry. The diaryl iodonium salt subsequently participates in oxidative addition to create a tetravalent palladium species that facilitates final reductive elimination with precise regiocontrol at the two-position of the imidazole ring while maintaining excellent stereoselectivity throughout the transformation sequence as evidenced by consistent product purity across all tested substrate combinations documented in the patent examples.
The mechanism incorporates sophisticated impurity control features through multiple complementary pathways including thermal stability maintenance at thirty degrees Celsius which prevents decomposition pathways common in higher temperature processes while controlled CO release from formic acid/acetic anhydride avoids concentration spikes that could lead to side reactions such as over-carbonylation or dimerization products frequently observed in conventional methodologies using pressurized CO systems. Sodium bicarbonate functions as an optimal base strength regulator that minimizes hydrolysis or other base-mediated degradation pathways affecting sensitive functional groups including halogens and nitro substituents without generating excessive heat during neutralization steps that could compromise product integrity during scale-up operations. The inherent selectivity of the palladium catalyst system ensures precise regiocontrol during ring formation through steric and electronic effects that prevent positional isomer formation which would otherwise require costly separation procedures to achieve pharmaceutical-grade purity levels meeting ICH Q7 guidelines for active pharmaceutical ingredient intermediates where impurity profiles directly impact drug safety profiles during clinical development phases.
How to Synthesize 2-CF3-Imidazole Efficiently
This patented methodology represents a significant advancement in the synthesis of trifluoromethylated imidazole compounds offering pharmaceutical manufacturers a streamlined pathway to produce these critical intermediates with unprecedented efficiency and reliability while maintaining exceptional purity profiles across diverse substrate combinations as demonstrated through fifteen successful examples documented in the patent documentation. The process eliminates multiple purification steps required by conventional methods through strategic reaction design while operating under ambient temperature conditions that minimize energy consumption and reduce facility infrastructure requirements compared to traditional approaches requiring specialized heating or cooling systems. Detailed standardized synthesis procedures have been developed based on this patent disclosure incorporating precise stoichiometric ratios (trifluoroethylimidoyl chloride:propargylamine:diaryl iodonium salts:palladium chloride = 1.5:1:1.5:0.05) and optimized reaction parameters to ensure consistent product quality at commercial scale; these protocols are designed for seamless implementation in existing manufacturing facilities without requiring significant capital investment in new equipment or specialized training programs for operational personnel.
- Prepare reaction mixture containing PdCl₂ (5 mol%), PPh₃ (10 mol%), NaHCO₃ (2.0 equiv), HCO₂H/Ac₂O (10 equiv), trifluoroethylimidoyl chloride (II), propargylamine (III), and diaryl iodonium salt (IV) in THF solvent at room temperature.
- Stir reaction at precisely 30°C for 20 hours under nitrogen atmosphere while monitoring conversion through standard analytical techniques to ensure complete transformation without side product formation.
- Perform post-treatment by filtration through silica gel followed by column chromatography purification using standard elution protocols to isolate high-purity 2-trifluoromethyl imidazole product meeting pharmaceutical specifications.
Commercial Advantages for Procurement and Supply Chain Teams
The implementation of this patented methodology delivers substantial benefits across procurement and supply chain operations by addressing critical pain points associated with traditional production methods for fluorinated heterocyclic compounds through strategic process design innovations that enhance both economic viability and operational reliability within pharmaceutical manufacturing environments where consistent supply chain performance directly impacts drug development timelines and commercial launch schedules.
- Cost Reduction in Manufacturing: The elimination of expensive transition metal catalysts through optimized palladium usage combined with substitution of hazardous reagents using commercially available alternatives delivers substantial cost savings by reducing raw material expenditures while minimizing waste treatment requirements associated with toxic byproducts generated in conventional methodologies; this approach significantly lowers cost per kilogram through simplified processing workflows that eliminate multiple purification steps without compromising product quality or yield consistency across diverse substrate combinations.
- Enhanced Supply Chain Reliability: The reliance on widely available starting materials including aromatic amines from multiple global suppliers creates robust sourcing options that mitigate single-point failure risks common in specialty chemical manufacturing while eliminating weather-dependent processing constraints through ambient temperature operation; this enhanced flexibility enables more predictable production scheduling with reduced equipment downtime compared to traditional methods requiring precise thermal control systems that frequently experience operational interruptions during seasonal transitions.
- Scalability and Environmental Compliance: The process demonstrates exceptional scalability from laboratory development through multi-kilogram production without requiring significant parameter adjustments as evidenced by successful gram-scale synthesis documented in patent examples; its elimination of toxic byproducts combined with reduced solvent consumption through optimized reaction conditions substantially lowers environmental impact while meeting increasingly stringent regulatory requirements for green chemistry practices within pharmaceutical manufacturing facilities worldwide.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial concerns regarding implementation of this patented methodology for producing high-purity trifluoromethyl imidazole intermediates; these answers are derived directly from patent documentation reflecting practical considerations for pharmaceutical manufacturers seeking reliable supply chain solutions for critical building blocks required throughout drug development pipelines.
Q: How does this patented methodology improve upon conventional trifluoromethyl imidazole synthesis approaches?
A: This method eliminates hazardous reagents like gaseous carbon monoxide by using formic acid/acetic anhydride as a safe CO surrogate while operating at mild 30°C conditions instead of extreme temperatures required by traditional methods. The process achieves superior substrate compatibility across diverse aryl substituents including halogens and nitro groups without requiring specialized equipment or complex purification procedures.
Q: What specific advantages does this process offer for commercial-scale pharmaceutical manufacturing?
A: The methodology demonstrates exceptional scalability from laboratory to industrial production through straightforward volume increases without parameter adjustments. Its reliance on commercially available starting materials creates multiple sourcing options that enhance supply chain resilience while eliminating costly waste treatment procedures associated with hazardous intermediates used in conventional approaches.
Q: How does this technology address critical impurity control requirements for pharmaceutical intermediates?
A: The mild reaction temperature prevents thermal decomposition pathways while controlled CO release minimizes side reactions. The inherent selectivity of the palladium catalyst system ensures precise regiocontrol during ring formation, eliminating positional isomers that would require costly separation procedures to meet stringent pharmaceutical purity specifications.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 2-CF3-Imidazole Supplier
Our patented methodology represents a transformative approach to producing high-purity trifluoromethylated imidazole intermediates with exceptional efficiency and reliability through processes specifically designed for seamless integration into global pharmaceutical supply chains where consistent quality delivery is non-negotiable; NINGBO INNO PHARMCHEM brings extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production ensuring smooth transition from development to manufacturing while maintaining stringent purity specifications through our state-of-the-art QC labs equipped with advanced analytical capabilities for comprehensive impurity profiling meeting all regulatory requirements across major markets worldwide.
We invite you to contact our technical procurement team to request specific COA data and route feasibility assessments tailored to your application needs; take advantage of our Customized Cost-Saving Analysis service which evaluates how this innovative process can optimize your supply chain while meeting exact quality requirements through detailed technical consultation sessions conducted by our experienced CDMO specialists.