Advanced One-Pot Synthesis of 2-Trifluoromethyl Quinazolinones for Scalable Pharmaceutical Production
Advanced One-Pot Synthesis of 2-Trifluoromethyl Quinazolinones for Scalable Pharmaceutical Production
The pharmaceutical and agrochemical industries are constantly seeking robust synthetic routes to access complex heterocyclic scaffolds that serve as the backbone for next-generation therapeutics. A significant breakthrough in this domain is documented in Chinese Patent CN112480015B, which discloses a highly efficient multi-component one-pot method for synthesizing 2-trifluoromethyl substituted quinazolinones. This class of compounds is critically important due to the prevalence of the quinazolinone core in numerous bioactive molecules, including well-known agents such as methaqualone and various antifungal and anticancer drugs. The introduction of a trifluoromethyl group further enhances these molecules by improving their metabolic stability, lipophilicity, and bioavailability, making them prime candidates for lead optimization in drug discovery programs. The patented methodology represents a paradigm shift from traditional multi-step sequences to a streamlined, catalytic cascade that maximizes atom economy and operational simplicity.
For research and development directors overseeing pipeline expansion, the ability to rapidly generate diverse libraries of fluorinated heterocycles is invaluable. The technique described in CN112480015B utilizes readily available nitro compounds and trifluoroethylimidoyl chlorides as starting materials, bypassing the need for costly and unstable intermediates often required in classical approaches. By leveraging a palladium-catalyzed carbonylation cascade, this process achieves high reaction efficiency and excellent substrate compatibility. The strategic integration of molybdenum hexacarbonyl as a solid carbon monoxide surrogate not only mitigates the safety hazards associated with handling high-pressure CO gas but also facilitates a smoother reaction profile. This technological advancement positions the synthesis of 2-trifluoromethyl quinazolinones as a viable option for both early-stage medicinal chemistry and late-stage process development, offering a reliable pathway for producing high-purity pharmaceutical intermediates.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the construction of the quinazolinone ring system has been fraught with synthetic challenges that hinder efficient large-scale manufacturing. Traditional methods often rely on the cyclization of anthranilic acid derivatives or the condensation of o-aminobenzamides, which frequently require harsh reaction conditions, strong acids or bases, and elevated temperatures that can degrade sensitive functional groups. Furthermore, many established protocols necessitate the use of high-pressure carbon monoxide atmospheres to introduce carbonyl functionality, posing significant safety risks and requiring specialized, capital-intensive reactor equipment that is not universally available in standard laboratory or pilot plant settings. Another major bottleneck is the reliance on pre-activated substrates, such as 2-bromoformylanilines or acid anhydrides, which are often expensive, moisture-sensitive, and require additional synthetic steps to prepare, thereby increasing the overall cost of goods and extending the production timeline. These limitations collectively result in lower overall yields, narrower substrate scopes, and a higher environmental footprint due to increased waste generation.
The Novel Approach
In stark contrast to these legacy techniques, the novel approach detailed in the patent utilizes a sophisticated yet operationally simple palladium-catalyzed carbonylation cascade. This method ingeniously combines a trifluoroethylimidoyl chloride and a nitro compound in a single reaction vessel, eliminating the need for isolation of intermediates. The reaction proceeds through a tandem sequence where the nitro group is first reduced to an amine in situ, followed by coupling and cyclization, all driven by the synergistic action of a palladium catalyst and a molybdenum carbonyl source. As illustrated in the reaction scheme below, this one-pot strategy dramatically reduces the number of unit operations, solvent usage, and purification steps required.
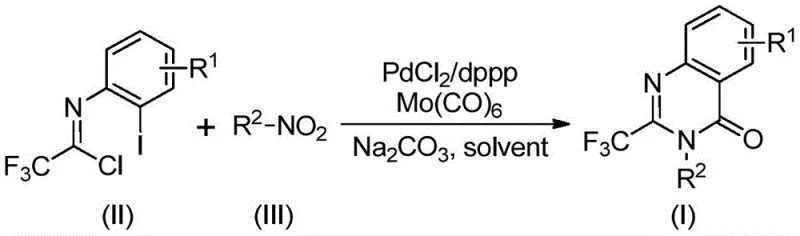
The use of nitro compounds as starting materials is particularly advantageous from a procurement perspective, as they are ubiquitous, inexpensive, and available in a vast array of substitution patterns. The reaction conditions are mild enough to tolerate a wide range of functional groups, including halogens, alkyls, and alkoxy groups, without the need for protecting group strategies. This robustness allows chemists to access complex molecular architectures directly from simple building blocks, significantly accelerating the timeline from concept to compound. Moreover, the replacement of gaseous CO with solid Mo(CO)6 simplifies the engineering controls required for the reaction, making the process inherently safer and more adaptable to standard glassware or stainless steel reactors found in most contract development and manufacturing organizations.
Mechanistic Insights into Pd-Catalyzed Carbonylation Cascade
Understanding the mechanistic underpinnings of this transformation is crucial for optimizing reaction parameters and troubleshooting potential scale-up issues. The proposed catalytic cycle begins with the reduction of the nitro compound to the corresponding amine by molybdenum hexacarbonyl, which serves as both a reducing agent and a carbon monoxide source. Once the amine is generated, it undergoes a base-promoted nucleophilic attack on the trifluoroethylimidoyl chloride to form a trifluoroacetamidine intermediate. Concurrently, the palladium catalyst, coordinated by the bidentate phosphine ligand dppp, inserts into the carbon-iodine bond of the imidoyl chloride derivative, generating a reactive organopalladium species. This step is critical for activating the substrate towards subsequent carbonylation.
As the reaction temperature is maintained at 120 °C, the molybdenum hexacarbonyl thermally decomposes to release carbon monoxide in a controlled manner. This in situ generated CO then inserts into the carbon-palladium bond to form an acyl-palladium intermediate. The proximity of the nitrogen atom within the acetamidine moiety facilitates an intramolecular nucleophilic attack on the acyl carbon, leading to the formation of a seven-membered palladacycle. Finally, reductive elimination from this cyclic intermediate releases the desired 2-trifluoromethyl substituted quinazolinone product and regenerates the active palladium(0) catalyst to continue the cycle. This intricate interplay between reduction, coupling, and cyclization ensures high atom efficiency and minimizes the formation of side products.
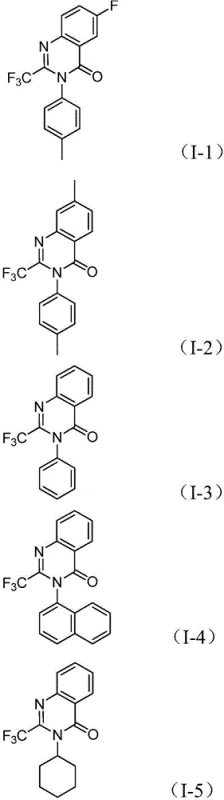
The versatility of this mechanism is evidenced by the broad substrate scope demonstrated in the patent examples. Various substituents on the aromatic ring of the nitro compound, such as methyl, fluoro, chloro, and bromo groups, are well-tolerated, yielding the corresponding products in high isolated yields ranging from 69% to 96%. Similarly, the nature of the R2 group on the nitro compound can be varied from simple phenyl rings to naphthyl and cyclohexyl groups, showcasing the method's adaptability for synthesizing diverse chemical spaces. The structural diversity achievable through this route is highlighted by the specific examples provided, which include compounds with varying electronic and steric properties, confirming the robustness of the catalytic system.
How to Synthesize 2-Trifluoromethyl Quinazolinones Efficiently
Implementing this synthesis in a laboratory or pilot plant setting requires careful attention to reagent stoichiometry and reaction conditions to maximize yield and purity. The protocol is designed to be user-friendly, utilizing standard Schlenk techniques or sealed tube reactions to maintain an inert atmosphere, although the use of solid CO sources reduces the sensitivity to air compared to traditional carbonylations. The following guide outlines the generalized procedure derived from the patent's preferred embodiments, ensuring reproducibility and high-quality output for your research or production needs. For a detailed, step-by-step standardized operating procedure tailored to your specific facility capabilities, please refer to the technical documentation provided below.
- Charge a reaction vessel with palladium chloride (5 mol %), dppp ligand (10 mol %), sodium carbonate (2.0 equiv), Mo(CO)6 (2.0 equiv), trifluoroethylimidoyl chloride, and the specific nitro compound substrate in 1,4-dioxane solvent.
- Heat the reaction mixture to 120 °C and maintain stirring for a duration of 16 to 30 hours to ensure complete conversion of the starting materials into the desired heterocyclic product.
- Upon completion, filter the mixture, adsorb the crude product onto silica gel, and purify via column chromatography to isolate the high-purity 2-trifluoromethyl substituted quinazolinone.
Commercial Advantages for Procurement and Supply Chain Teams
From a supply chain and procurement standpoint, the adoption of this novel synthetic route offers compelling economic and logistical benefits that directly impact the bottom line. The primary driver for cost reduction lies in the raw material selection; nitro compounds and trifluoroethylimidoyl chlorides are commodity chemicals available from multiple global suppliers, ensuring a competitive pricing landscape and reducing the risk of supply bottlenecks. Unlike methods requiring custom-synthesized precursors or rare earth catalysts, this process relies on established palladium chemistry with ligands that are commercially off-the-shelf. The elimination of high-pressure gas infrastructure further reduces capital expenditure requirements, allowing for production in existing facilities without major retrofitting. Additionally, the simplified workup procedure involving filtration and standard chromatography minimizes solvent consumption and waste disposal costs, contributing to a more sustainable and cost-effective manufacturing process.
- Cost Reduction in Manufacturing: The economic viability of this process is significantly enhanced by the use of inexpensive starting materials and the avoidance of complex pre-activation steps. By consolidating multiple synthetic transformations into a single pot, the method drastically reduces labor hours, energy consumption, and solvent usage associated with intermediate isolations. The high reaction efficiency and yields reported in the patent data mean that less raw material is wasted, directly lowering the cost per kilogram of the final active pharmaceutical ingredient intermediate. Furthermore, the ability to use standard heating equipment rather than specialized high-pressure autoclaves lowers the barrier to entry for contract manufacturers, fostering a more competitive supply market.
- Enhanced Supply Chain Reliability: Supply chain resilience is bolstered by the widespread availability of the key reagents. Nitro compounds are produced on a massive scale for various industries, ensuring a stable and continuous supply stream even during market fluctuations. The robustness of the reaction conditions means that minor variations in raw material quality are less likely to cause batch failures, reducing the risk of production delays. This reliability is crucial for maintaining consistent inventory levels and meeting tight delivery schedules for downstream drug development projects. The scalability of the process from gram to potentially tonnage scales ensures that supply can grow in tandem with clinical demand, preventing shortages during critical phases of drug commercialization.
- Scalability and Environmental Compliance: The environmental profile of this synthesis aligns well with modern green chemistry principles and regulatory expectations. The use of Mo(CO)6 as a solid CO source eliminates the safety hazards and regulatory burdens associated with storing and transporting toxic carbon monoxide gas cylinders. The reaction generates fewer byproducts compared to traditional methods, simplifying effluent treatment and reducing the environmental footprint of the manufacturing site. The high atom economy of the cascade reaction ensures that a greater proportion of the input mass is converted into the desired product, minimizing waste generation. These factors collectively facilitate easier regulatory approval and support the long-term sustainability goals of pharmaceutical companies aiming to reduce their carbon footprint.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis technology. These answers are derived directly from the experimental data and specifications outlined in the patent literature, providing a factual basis for decision-making. Understanding these nuances helps stakeholders evaluate the feasibility of integrating this method into their existing workflows and assess its potential impact on project timelines and budgets.
Q: What are the primary advantages of this one-pot method over traditional quinazolinone synthesis?
A: This method eliminates the need for high-pressure carbon monoxide gas and expensive pre-activated substrates. By using inexpensive nitro compounds and solid Mo(CO)6 as a CO source, it significantly simplifies the operational setup and improves safety profiles for industrial scale-up.
Q: What is the substrate scope for the R1 and R2 groups in this synthesis?
A: The process demonstrates excellent functional group tolerance. R1 can be hydrogen, alkyl, halogens, or trifluoromethyl groups, while R2 accommodates alkyl, cycloalkyl, and various substituted aryl groups, allowing for the creation of diverse chemical libraries for drug discovery.
Q: Is this synthesis suitable for large-scale commercial production?
A: Yes, the patent explicitly mentions scalability to the gram level with potential for industrial application. The use of standard organic solvents like dioxane and commercially available catalysts supports a straightforward transition from laboratory to pilot plant manufacturing.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 2-Trifluoromethyl Quinazolinone Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical role that advanced synthetic methodologies play in accelerating drug discovery and development. Our team of expert chemists has extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that promising laboratory results translate seamlessly into industrial reality. We are committed to delivering high-purity pharmaceutical intermediates that meet stringent purity specifications, supported by our rigorous QC labs equipped with state-of-the-art analytical instrumentation. Whether you require custom synthesis of novel quinazolinone derivatives or scale-up of existing processes, our facility is designed to handle complex chemistry with precision and reliability.
We invite you to collaborate with us to leverage this cutting-edge technology for your next project. Our technical procurement team is ready to provide a Customized Cost-Saving Analysis tailored to your specific volume requirements and quality standards. Please contact us today to request specific COA data for our catalog compounds or to discuss route feasibility assessments for your proprietary molecules. Together, we can optimize your supply chain and bring life-saving therapies to market faster and more efficiently.