Advanced Palladium-Catalyzed Synthesis of Carbonyl-Bridged Biheterocyclic Compounds for Pharma
Advanced Palladium-Catalyzed Synthesis of Carbonyl-Bridged Biheterocyclic Compounds for Pharma
The pharmaceutical industry continuously seeks efficient pathways to construct complex heterocyclic scaffolds that serve as the core backbone for novel therapeutic agents. Patent CN115353511A discloses a groundbreaking multi-component method for synthesizing carbonyl-bridged biheterocyclic compounds, specifically targeting the fusion of indolinone and imidazole motifs. This technology represents a significant leap forward in organic synthesis, addressing the critical need for streamlined processes that can deliver high-purity pharmaceutical intermediates without the logistical burdens of hazardous reagents. By leveraging a transition metal palladium-catalyzed carbonylation cascade, this method enables the one-pot assembly of diversified double heterocyclic compounds containing trifluoromethyl and carbonyl functionalities. For R&D directors and procurement specialists, this innovation offers a compelling value proposition: it simplifies the supply chain by utilizing cheap, readily available starting materials while eliminating the need for toxic carbon monoxide gas cylinders, thereby aligning with modern green chemistry principles and stringent safety regulations in chemical manufacturing facilities.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the construction of biheterocyclic systems has relied on laborious multi-step sequences that often suffer from poor atom economy and low overall yields. Traditional approaches typically involve the direct coupling of two pre-formed heterocyclic substrates, which requires the prior synthesis and purification of each individual ring system, drastically increasing production time and waste generation. Alternatively, oxidative cyclization reactions using activated methyl-substituted heterocycles often demand harsh oxidizing conditions that are incompatible with sensitive functional groups, limiting the structural diversity of the final library. Furthermore, conventional carbonylation strategies to introduce the crucial carbonyl bridge frequently necessitate the use of high-pressure carbon monoxide gas, posing severe safety risks and requiring specialized, expensive high-pressure reactors that are not universally available in standard pilot plants. These legacy methods create significant bottlenecks in the development of new drug candidates, particularly when rapid iteration of chemical structures is required to optimize biological activity profiles.
The Novel Approach
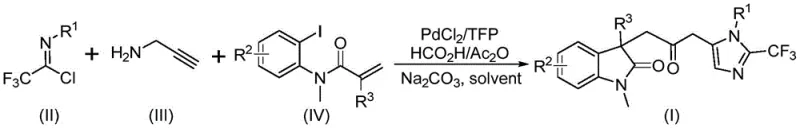
In stark contrast, the methodology described in patent CN115353511A introduces a highly efficient, one-pot multicomponent reaction that constructs the entire biheterocyclic framework in a single operational step. This novel approach utilizes a palladium catalyst system to orchestrate a tandem sequence involving C-I bond activation, intramolecular Heck cyclization, and carbonylation, all occurring under remarkably mild conditions at 30°C. The strategic use of a formic acid and acetic anhydride mixture serves as a safe, liquid surrogate for carbon monoxide, releasing the gas in situ exactly where it is needed within the catalytic cycle. This eliminates the hazards associated with handling compressed CO gas and allows the reaction to proceed in standard glassware, such as Schlenk tubes, making it accessible for both laboratory discovery and potential scale-up. The versatility of this system is further enhanced by its broad substrate tolerance, enabling the incorporation of various electronic and steric environments on the aromatic rings without compromising reaction efficiency.
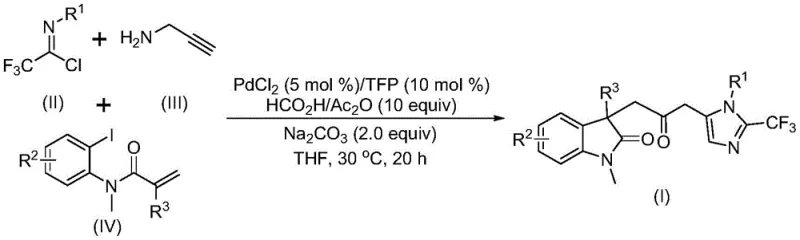
The general reaction scheme illustrates the convergence of three distinct building blocks into a single, complex molecular architecture. This convergent strategy is inherently superior to linear synthesis routes because it maximizes the speed of structure generation and minimizes the accumulation of intermediate impurities. For a reliable pharmaceutical intermediate supplier, adopting such a convergent multicomponent reaction translates directly into reduced manufacturing costs and shorter lead times for clients seeking custom synthesis services. The ability to introduce a trifluoromethyl group simultaneously with the ring closure is particularly valuable, as fluorine atoms are ubiquitous in modern medicinal chemistry for their ability to enhance metabolic stability and membrane permeability of drug candidates.
Mechanistic Insights into Pd-Catalyzed Carbonylation Cascade
Understanding the mechanistic underpinnings of this transformation is crucial for R&D teams aiming to optimize the process for specific analogs. The reaction is initiated by the oxidative addition of a zero-valent palladium species into the carbon-iodine bond of the acrylamide derivative, generating a reactive aryl-palladium intermediate. This species subsequently undergoes an intramolecular Heck-type insertion into the pendant alkene, forming a five-membered indolinone ring and a divalent alkyl-palladium intermediate. It is at this stage that the in situ generated carbon monoxide inserts into the palladium-carbon bond, creating an acyl-palladium complex that serves as the electrophilic center for the subsequent cyclization. Concurrently, in a separate but synchronized pathway, the base-promoted reaction between trifluoroethylimidoyl chloride and propargylamine generates a trifluoroacetamidine intermediate, which then undergoes isomerization to become nucleophilic enough to attack the acyl-palladium species. This intricate dance of bond formations culminates in a final reductive elimination or cyclization step that releases the product and regenerates the active palladium catalyst, closing the catalytic loop.


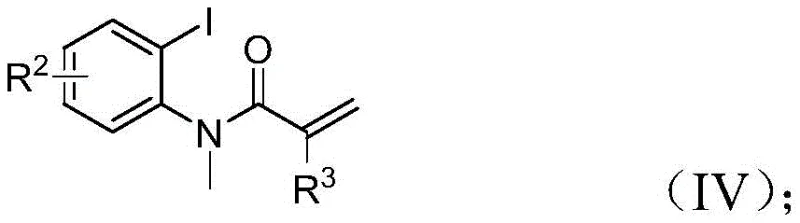
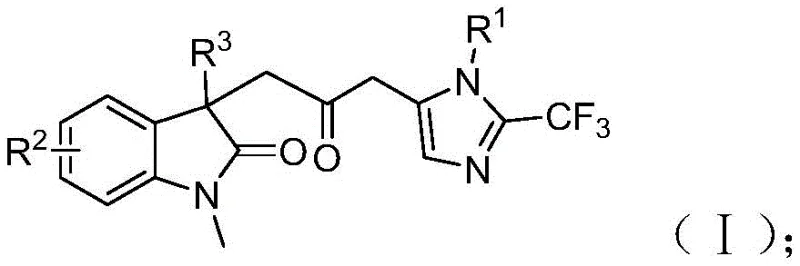
The specific structures of the reactants play a pivotal role in driving this mechanism forward. The trifluoroethylimidoyl chloride acts as the source of the trifluoromethyl-imidazole moiety, while propargylamine provides the nitrogen and carbon atoms necessary for the second heterocyclic ring. The acrylamide derivative, bearing the iodine handle, serves as the scaffold for the indolinone formation. The compatibility of this mechanism with various substituents on the aromatic rings (R1, R2, and R3) suggests that the electronic properties of the substrates do not severely inhibit the catalytic turnover. For instance, electron-withdrawing groups like nitro or trifluoromethyl, as well as electron-donating groups like methoxy or methyl, are all tolerated, indicating a robust catalytic system that is less sensitive to substrate electronics than many traditional cross-coupling reactions. This robustness is a key factor in ensuring consistent quality and yield across a diverse range of target molecules.
How to Synthesize Carbonyl-Bridged Biheterocyclic Compounds Efficiently
Executing this synthesis requires careful attention to the stoichiometry and the order of addition to ensure optimal conversion. The patent specifies a preferred molar ratio where the trifluoroethylimidoyl chloride is the limiting reagent, while propargylamine and the acrylamide derivative are used in slight excess to drive the equilibrium towards the product. The reaction is typically conducted in an aprotic organic solvent such as tetrahydrofuran (THF), which effectively dissolves all organic components and stabilizes the palladium intermediates. Sodium carbonate acts as the base to neutralize the hydrochloric acid byproduct generated during the amidine formation, preventing catalyst poisoning. The reaction temperature is maintained at a mild 30°C, which is sufficient to activate the catalyst without promoting decomposition of the sensitive intermediates. Following the reaction period of 12 to 20 hours, the crude mixture is filtered to remove inorganic salts and palladium black, and the product is isolated via standard silica gel column chromatography.
- Combine palladium chloride, TFP ligand, sodium carbonate, and the CO source mixture (formic acid/acetic anhydride) in an organic solvent like THF.
- Add the three key substrates: trifluoroethylimidoyl chloride, propargylamine, and the acrylamide derivative to the reaction vessel.
- Stir the mixture at 30°C for 12 to 20 hours, then filter and purify via column chromatography to isolate the target biheterocycle.
Commercial Advantages for Procurement and Supply Chain Teams
From a commercial perspective, this patented methodology offers substantial advantages that directly impact the bottom line for chemical procurement and supply chain management. The primary driver for cost reduction lies in the simplicity of the operation and the nature of the raw materials. By consolidating multiple synthetic steps into a single pot, the process eliminates the need for intermediate isolation, purification, and drying, which are traditionally the most time-consuming and solvent-intensive parts of chemical manufacturing. Furthermore, the avoidance of high-pressure carbon monoxide equipment removes a significant capital expenditure barrier and reduces the regulatory compliance burden associated with handling toxic gases. This simplification of the process infrastructure allows for more flexible manufacturing scheduling and lower overhead costs per kilogram of product produced.
- Cost Reduction in Manufacturing: The economic benefits of this route are derived from the use of commodity chemicals as starting materials. Trifluoroethylimidoyl chloride, propargylamine, and acrylamide derivatives are commercially available in bulk quantities at competitive prices, ensuring a stable and cost-effective supply chain. The high atom economy of the multicomponent reaction means that a larger proportion of the raw material mass is incorporated into the final product, reducing waste disposal costs. Additionally, the mild reaction conditions (30°C) significantly lower energy consumption compared to processes requiring reflux or cryogenic temperatures, contributing to a smaller carbon footprint and reduced utility expenses for the manufacturing facility.
- Enhanced Supply Chain Reliability: Supply chain resilience is bolstered by the broad availability of the requisite reagents. Unlike specialized organometallic reagents that may have long lead times or single-source suppliers, the components of this reaction are standard catalog items for major chemical distributors. This diversity of supply sources mitigates the risk of production delays due to raw material shortages. Moreover, the robustness of the reaction against moisture and air (to a reasonable extent, given the use of Schlenk techniques) simplifies logistics and storage requirements, allowing for safer and more economical transportation of materials to the production site without the need for extreme environmental controls.
- Scalability and Environmental Compliance: The transition from laboratory scale to commercial production is facilitated by the homogeneous nature of the reaction mixture and the absence of hazardous gas feeds. The patent demonstrates successful gram-scale synthesis, providing a clear proof-of-concept for kilogram and ton-scale operations. From an environmental standpoint, the use of formic acid as a CO surrogate generates water and carbon dioxide as benign byproducts, rather than heavy metal waste or toxic effluents. This aligns with increasingly strict global environmental regulations, making it easier to obtain permits for large-scale production and reducing the long-term liability associated with hazardous waste management.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis technology. These answers are derived directly from the experimental data and specifications outlined in the patent documentation, providing a factual basis for decision-making. Understanding these details helps stakeholders assess the feasibility of integrating this method into their existing development pipelines or manufacturing portfolios.
Q: What are the safety advantages of this carbonylation method compared to traditional methods?
A: Unlike traditional carbonylation which requires toxic and high-pressure carbon monoxide gas, this patent utilizes a formic acid and acetic anhydride mixture to generate CO in situ under mild conditions, significantly enhancing operational safety.
Q: What is the substrate scope for the R1 and R3 groups in this synthesis?
A: The method demonstrates excellent compatibility with various substituents. R1 can be alkyl or substituted aryl groups (including halogens, nitro, and trifluoromethyl), while R3 accommodates alkyl, phenyl, or benzyl groups, allowing for diverse library synthesis.
Q: Is this process suitable for large-scale industrial production?
A: Yes, the patent explicitly mentions that the reaction conditions are mild (30°C) and the procedure has been successfully expanded to gram-scale reactions, indicating strong potential for commercial scale-up with standard processing equipment.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Carbonyl-Bridged Biheterocyclic Compounds Supplier
At NINGBO INNO PHARMCHEM, we recognize the transformative potential of advanced catalytic methodologies like the one described in patent CN115353511A for accelerating drug discovery programs. As a leading CDMO partner, we possess the technical expertise and infrastructure to translate these innovative academic protocols into robust, GMP-compliant manufacturing processes. Our team has extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your project can seamlessly transition from milligram-scale screening to full-scale commercial supply. We maintain stringent purity specifications and operate rigorous QC labs equipped with state-of-the-art analytical instrumentation to guarantee that every batch of carbonyl-bridged biheterocyclic compounds meets the highest quality standards required by the global pharmaceutical industry.
We invite you to collaborate with us to leverage this cutting-edge synthesis technology for your next project. Our technical procurement team is ready to provide a Customized Cost-Saving Analysis tailored to your specific volume requirements and timeline. By partnering with us, you gain access to our deep reservoir of process chemistry knowledge and our commitment to delivering high-purity intermediates with unmatched reliability. Please contact us today to request specific COA data for similar scaffolds or to discuss route feasibility assessments for your target molecules, and let us help you bring your novel therapeutics to market faster and more efficiently.