Scalable Metal-Free Synthesis of Trifluoromethyl Triazoles for Advanced Pharmaceutical Intermediates
Scalable Metal-Free Synthesis of Trifluoromethyl Triazoles for Advanced Pharmaceutical Intermediates
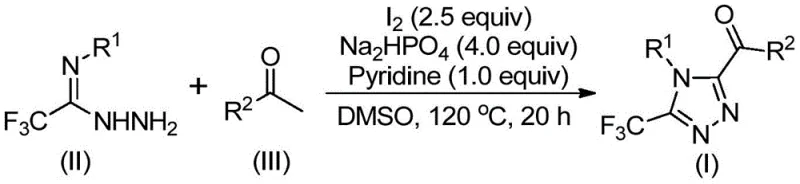
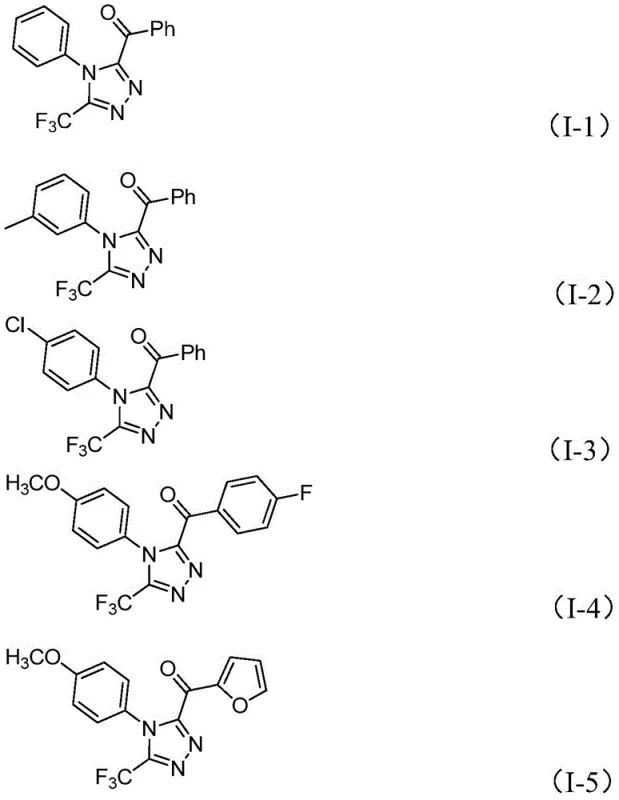
The pharmaceutical industry continuously seeks robust synthetic routes for nitrogen-containing heterocycles, particularly 1,2,4-triazoles, which serve as critical scaffolds in numerous bioactive molecules including antiviral and antidiabetic agents. Patent CN113105402B introduces a groundbreaking preparation method for 3,4,5-trisubstituted 1,2,4-triazole compounds that addresses long-standing challenges in heterocyclic synthesis. This technology leverages a non-metallic iodine-promoted cascade reaction, utilizing readily available aryl ethyl ketones and trifluoroethylimide hydrazides as starting materials. By eliminating the need for expensive transition metal catalysts and严苛 anhydrous conditions, this innovation represents a significant leap forward in process chemistry efficiency. For R&D directors and procurement specialists, this patent offers a viable pathway to high-purity intermediates with a drastically simplified impurity profile, ensuring compliance with stringent global regulatory standards for active pharmaceutical ingredients.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Traditionally, the construction of polysubstituted 1,2,4-triazole rings, especially those bearing both trifluoromethyl and acyl groups, has relied on complex multi-step sequences or the use of precious metal catalysts. Conventional methodologies often necessitate rigorous exclusion of moisture and oxygen, requiring specialized equipment and inert gas manifolds that increase capital expenditure and operational complexity. Furthermore, the reliance on heavy metal catalysts introduces significant downstream processing burdens, as residual metal levels must be reduced to parts-per-million (ppm) levels to meet ICH Q3D guidelines for elemental impurities. These purification steps not only extend lead times but also result in substantial yield losses during chromatography or recrystallization, thereby inflating the overall cost of goods sold (COGS) for the final API intermediate.
The Novel Approach
In stark contrast, the novel approach detailed in the patent utilizes a tandem iodination and Kornblum oxidation strategy mediated by elemental iodine in dimethyl sulfoxide (DMSO). This method transforms simple aryl ethyl ketones into reactive aryl diketones in situ, which subsequently undergo condensation and cyclization with trifluoroethylimide hydrazides. The process operates under ambient atmospheric conditions, removing the necessity for gloveboxes or Schlenk lines. By employing cheap, commodity chemicals like iodine and sodium dihydrogen phosphate, the method significantly lowers the raw material entry barrier. This shift from noble metal catalysis to halogen-mediated organic synthesis provides a greener, more economical alternative that maintains high functional group tolerance, allowing for the diverse substitution patterns required in modern drug discovery campaigns.
Mechanistic Insights into Iodine-Promoted Cyclization
The mechanistic pathway of this transformation is a sophisticated interplay of oxidation and condensation events driven by the unique properties of the iodine-DMSO system. Initially, the aryl ethyl ketone undergoes alpha-iodination followed by Kornblum oxidation, where DMSO acts as the oxygen donor to generate an alpha-dicarbonyl intermediate. This highly reactive species then condenses with the nucleophilic nitrogen of the trifluoroethylimide hydrazide to form a hydrazone intermediate. The presence of pyridine and sodium dihydrogen phosphate serves a dual purpose: buffering the acidic byproducts generated during iodination and facilitating the subsequent intramolecular cyclization. The final ring closure yields the stable 1,2,4-triazole core with the trifluoromethyl group positioned at the 3-position and the acyl group at the 5-position, a regioselectivity that is difficult to achieve with other methods.
From an impurity control perspective, this mechanism is advantageous because the reaction intermediates are transient and convert efficiently to the final product under the optimized thermal conditions of 110-130°C. The use of stoichiometric iodine ensures complete conversion of the starting ketone, minimizing the presence of unreacted starting materials in the crude mixture. Moreover, the absence of metal-ligand complexes eliminates a whole class of potential organometallic impurities that are notoriously difficult to characterize and remove. This clean reaction profile simplifies the work-up procedure, typically requiring only filtration and standard silica gel chromatography, which translates directly to higher throughput and reduced solvent consumption in a manufacturing setting.
How to Synthesize 3,4,5-Trisubstituted 1,2,4-Triazole Efficiently
The operational simplicity of this synthesis makes it highly attractive for process development teams aiming to scale up production rapidly. The protocol involves a straightforward two-stage heating sequence in a single reactor vessel, minimizing unit operations and transfer losses. Detailed standard operating procedures regarding reagent addition rates, temperature ramping, and quenching protocols are essential for maintaining reproducibility at the kilogram scale. The following guide outlines the critical steps derived from the patent examples to ensure optimal yield and purity.
- Mix aryl ethyl ketone and iodine in DMSO, heating to 90-110°C for 4-6 hours to initiate iodination and oxidation.
- Add additional iodine, sodium dihydrogen phosphate, pyridine, and trifluoroethylimide hydrazide to the reaction mixture.
- Heat the mixture to 110-130°C for 12-20 hours to complete the cyclization, followed by filtration and purification.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this iodine-promoted methodology offers tangible strategic benefits beyond mere technical feasibility. The primary advantage lies in the drastic simplification of the supply chain for raw materials; aryl ethyl ketones and elemental iodine are bulk commodities available from multiple global suppliers, mitigating the risk of single-source dependency. This abundance ensures price stability and consistent availability, which are critical factors for long-term production planning. Furthermore, the elimination of expensive ligands and metal catalysts removes a significant cost center from the bill of materials, directly improving the margin structure of the final intermediate.
- Cost Reduction in Manufacturing: The removal of heavy metal catalysts fundamentally alters the cost structure of the synthesis. Without the need for specialized metal scavengers or extensive purification trains to meet residual metal specifications, the downstream processing costs are significantly reduced. The reaction utilizes DMSO, a low-cost polar aprotic solvent, further driving down operational expenses compared to processes requiring exotic solvents or cryogenic conditions. This economic efficiency allows for competitive pricing strategies when supplying high-volume pharmaceutical intermediates to generic and innovator drug companies alike.
- Enhanced Supply Chain Reliability: The robustness of the reaction conditions, specifically the tolerance to air and moisture, enhances supply chain reliability by reducing the sensitivity of the manufacturing process to environmental fluctuations. This resilience minimizes the risk of batch failures due to minor deviations in humidity or oxygen levels, ensuring consistent on-time delivery to customers. Additionally, the scalability of the method from gram to multi-kilogram levels means that supply can be ramped up quickly to meet surges in demand without requiring extensive re-engineering of the process or acquisition of specialized high-pressure equipment.
- Scalability and Environmental Compliance: From an environmental, health, and safety (EHS) perspective, this method aligns well with green chemistry principles by avoiding toxic heavy metals. This reduces the burden of hazardous waste disposal and lowers the environmental compliance costs associated with wastewater treatment. The ability to run the reaction at moderate temperatures (up to 130°C) without high pressure also reduces energy consumption and safety risks related to reactor containment. These factors collectively contribute to a more sustainable manufacturing footprint, which is increasingly a key criterion for selection by top-tier multinational pharmaceutical corporations.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis technology. These insights are derived directly from the experimental data and claims within the patent documentation, providing clarity on the method's versatility and limitations. Understanding these nuances is vital for technical teams evaluating the feasibility of integrating this route into their existing manufacturing portfolios.
Q: Does this synthesis method require expensive heavy metal catalysts?
A: No, the process described in patent CN113105402B utilizes elemental iodine as a non-metallic promoter, completely avoiding the use of toxic heavy metal catalysts and simplifying downstream purification.
Q: What are the typical reaction conditions for this triazole formation?
A: The reaction proceeds in dimethyl sulfoxide (DMSO) via a two-stage heating process: initially at 90-110°C for oxidation, followed by 110-130°C for cyclization, without requiring strict anhydrous or oxygen-free environments.
Q: Is this method suitable for large-scale industrial production?
A: Yes, the patent explicitly states that the method is easily scalable from gram level to industrial production due to the use of cheap, commercially available raw materials and simple operational procedures.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 3,4,5-Trisubstituted 1,2,4-Triazole Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical role that efficient heterocyclic synthesis plays in accelerating drug development timelines. Our team of expert chemists has extensively evaluated the iodine-promoted pathway described in CN113105402B and validated its potential for commercial application. We possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that our clients receive a consistent supply of high-quality intermediates. Our state-of-the-art facilities are equipped with rigorous QC labs capable of verifying stringent purity specifications, guaranteeing that every batch meets the exacting standards required for GMP manufacturing of active pharmaceutical ingredients.
We invite potential partners to engage with our technical procurement team to discuss how this advanced synthesis route can be tailored to your specific project needs. By leveraging our expertise in process optimization, we can provide a Customized Cost-Saving Analysis that quantifies the economic benefits of switching to this metal-free methodology. Please contact us to request specific COA data for our triazole library or to schedule a consultation regarding route feasibility assessments for your next-generation therapeutic candidates.