Advanced Palladium-Catalyzed Synthesis of 2-Trifluoromethyl Quinazolinones for Pharmaceutical Applications
The pharmaceutical and fine chemical industries are constantly seeking robust methodologies to access nitrogen-containing heterocycles, particularly those bearing fluorinated motifs which are known to enhance metabolic stability and bioavailability. Patent CN113045503B discloses a groundbreaking preparation method for 2-trifluoromethyl substituted quinazolinone compounds, a structural scaffold prevalent in numerous bioactive agents ranging from antifungals to anticancer drugs. This technology represents a significant leap forward in organic synthesis, offering a transition metal palladium-catalyzed carbonylation cascade that bypasses the limitations of classical cyclization routes. By utilizing readily available trifluoroethylimidoyl chloride and various amines as starting materials, this process achieves exceptional reaction efficiency and substrate compatibility. For R&D directors and procurement specialists alike, this innovation promises not only streamlined laboratory workflows but also a viable pathway for the cost reduction in pharmaceutical intermediates manufacturing, addressing the critical need for scalable and economically feasible production of high-value heterocyclic building blocks.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of 2-trifluoromethyl substituted quinazolinones has been plagued by significant operational and economic hurdles that hinder large-scale adoption. Traditional protocols often rely on the cyclization of anthranilamides with ethyl trifluoroacetate or trifluoroacetic anhydride, reactions that frequently demand harsh conditions, expensive pre-activated substrates, or the handling of unstable intermediates like trifluoroacetamides. Furthermore, alternative routes involving isatoic anhydrides or T3P-promoted cascade reactions often suffer from narrow substrate scopes and inconsistent yields, making them unreliable for diverse medicinal chemistry campaigns. These legacy methods typically require rigorous exclusion of moisture, elevated temperatures that degrade sensitive functional groups, and complex workup procedures to remove stubborn byproducts. Consequently, the supply chain for these critical intermediates has remained fragile, with high production costs and long lead times deterring the widespread exploration of trifluoromethylated quinazolinone libraries in drug discovery programs.
The Novel Approach
In stark contrast to these archaic techniques, the novel palladium-catalyzed carbonylation strategy described in the patent offers a streamlined, high-yielding alternative that fundamentally reshapes the synthetic landscape. This method employs a sophisticated catalytic system comprising palladium trifluoroacetate and triphenylphosphine, coupled with TFBen as a safe solid carbon monoxide surrogate, effectively eliminating the safety risks associated with high-pressure CO gas. The reaction proceeds smoothly in 1,4-dioxane at 110°C, tolerating a wide array of functional groups including halogens, alkyls, and ethers on both the aromatic ring and the amine component. As illustrated in the general reaction scheme below, the convergence of trifluoroethylimidoyl chloride and diverse amines leads directly to the target quinazolinone core with remarkable efficiency.
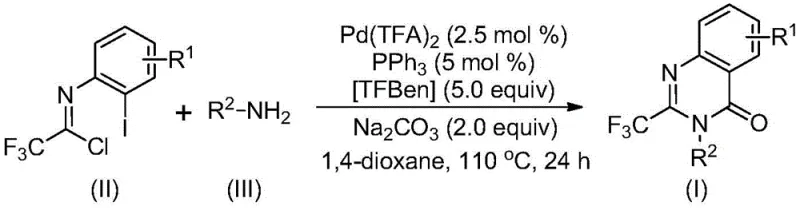
The versatility of this approach is evidenced by the successful synthesis of various derivatives, such as compounds I-1 through I-5, with isolated yields reaching as high as 98% in optimized examples. This robustness allows chemists to design and synthesize trifluoromethylquinazolinone compounds with different substitution patterns according to actual needs, significantly widening the practical applicability of the method for generating diverse chemical libraries without the burden of optimizing unique conditions for each substrate.
Mechanistic Insights into Pd-Catalyzed Carbonylation Cascade
Understanding the mechanistic underpinnings of this transformation is crucial for R&D teams aiming to replicate or modify the process for specific API intermediates. The reaction is believed to initiate with a base-promoted intermolecular carbon-nitrogen bond coupling between the amine and the trifluoroethylimidoyl chloride, generating a trifluoroacetamidine derivative in situ. Subsequently, the active palladium(0) species, generated from the precatalyst and ligand, undergoes oxidative addition into the carbon-iodine bond of the aromatic ring, forming a key divalent palladium intermediate. At this stage, the TFBen additive plays a pivotal role by thermally decomposing to release carbon monoxide, which then inserts into the carbon-palladium bond to form an acyl palladium species. This sequence avoids the need for external gas cylinders, enhancing safety and operational simplicity in standard laboratory or pilot plant settings.
Following CO insertion, the mechanism proceeds through an intramolecular cyclization facilitated by the base, which promotes the formation of a seven-membered ring palladium intermediate involving the nitrogen atom of the amidine moiety. The final step involves a reductive elimination that releases the desired 2-trifluoromethyl substituted quinazolinone compound and regenerates the active palladium catalyst to continue the cycle. This elegant cascade ensures high atom economy and minimizes the formation of side products, as the specific coordination environment created by the triphenylphosphine ligand directs the reaction pathway selectively towards the quinazolinone core. Such precise control over the catalytic cycle is essential for maintaining high purity specifications, reducing the burden on downstream purification processes like column chromatography, and ensuring that the final material meets the stringent quality standards required for pharmaceutical applications.
How to Synthesize 2-Trifluoromethyl Quinazolinones Efficiently
To implement this advanced synthetic route in your own facilities, it is essential to adhere to the optimized parameters regarding catalyst loading, solvent choice, and reaction time established in the patent data. The process is designed to be operationally simple, requiring standard Schlenk techniques or sealed vessel heating to maintain the necessary temperature of 110°C for a duration of 16 to 30 hours. The use of 1,4-dioxane as the solvent is preferred due to its ability to fully dissolve the organic substrates and facilitate the catalytic turnover, although other aprotic solvents may be evaluated for specific solubility challenges. Detailed standardized synthesis steps for replicating this high-efficiency transformation are provided in the guide below.
- Combine palladium trifluoroacetate, triphenylphosphine, TFBen (CO source), sodium carbonate, trifluoroethylimidoyl chloride, and amine in an organic solvent like 1,4-dioxane.
- Heat the reaction mixture to 110°C and maintain stirring for 16 to 30 hours to allow the carbonylation cascade and cyclization to proceed to completion.
- Upon completion, filter the mixture, mix with silica gel, and purify via column chromatography to isolate the high-purity 2-trifluoromethyl substituted quinazolinone product.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this novel synthesis method offers tangible strategic benefits that extend beyond mere chemical curiosity. The shift from expensive, unstable reagents to cheap, commercially available starting materials like trifluoroethylimidoyl chloride and common amines drastically simplifies the sourcing landscape. This accessibility ensures a more reliable quinazolinone derivative supplier network, as the raw materials are not subject to the same volatility or scarcity as specialized activated esters or anhydrides used in conventional methods. Furthermore, the elimination of high-pressure carbon monoxide gas removes a significant safety barrier and infrastructure cost, allowing for production in facilities that may not be equipped for hazardous gas handling, thereby expanding the pool of potential contract manufacturing organizations.
- Cost Reduction in Manufacturing: The economic impact of this process is profound, driven primarily by the use of inexpensive catalysts and reagents combined with high reaction yields that minimize raw material waste. By avoiding the need for pre-activation steps and utilizing a solid CO surrogate, the process significantly reduces the operational complexity and energy consumption associated with traditional high-pressure carbonylations. The high conversion rates observed, often exceeding 90%, mean that less starting material is required per kilogram of output, leading to substantial cost savings in the overall cost of goods sold (COGS) for these valuable intermediates.
- Enhanced Supply Chain Reliability: The robustness of the reaction conditions and the broad functional group tolerance contribute to a more predictable and stable supply chain. Since the method does not rely on sensitive reagents that degrade rapidly upon exposure to air or moisture, inventory management becomes more straightforward, and the risk of batch failures due to reagent quality issues is markedly reduced. This reliability is critical for maintaining continuous production schedules for key pharmaceutical intermediates, ensuring that downstream drug development timelines are not compromised by raw material shortages or inconsistent quality from suppliers.
- Scalability and Environmental Compliance: From a scale-up perspective, the protocol has been successfully demonstrated to be extendable to gram-scale synthesis without loss of efficiency, indicating strong potential for multi-kilogram or ton-scale production. The use of TFBen as a CO source inherently improves the environmental profile of the reaction by mitigating the risks of toxic gas leaks, aligning with increasingly strict global safety and environmental regulations. Additionally, the simplified post-treatment process, involving basic filtration and chromatography, reduces the volume of solvent waste and hazardous byproducts, facilitating easier compliance with waste disposal protocols and lowering the environmental footprint of the manufacturing process.
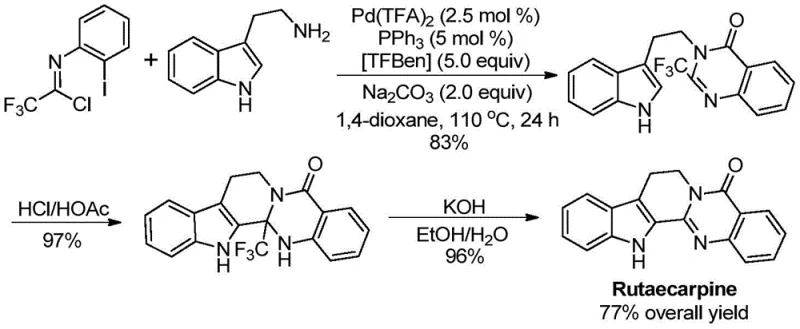
The practical utility of this method is perhaps best exemplified by its successful application in the total synthesis of the bioactive natural product Rutaecarpine. As shown in the reaction pathway below, the key quinazolinone intermediate was constructed efficiently, followed by simple acid-mediated cyclization and base treatment to afford the final drug molecule in a commendable 77% overall yield. This case study serves as a powerful proof of concept for the method's capability to handle complex molecular architectures relevant to the pharmaceutical industry.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this palladium-catalyzed synthesis technology. These answers are derived directly from the experimental data and beneficial effects reported in the patent documentation, providing clarity on reaction scope, safety, and scalability for potential partners and licensees.
Q: What are the primary advantages of this Pd-catalyzed method over conventional quinazolinone synthesis?
A: Unlike traditional methods requiring harsh conditions or unstable reagents like trifluoroacetamides, this novel approach utilizes cheap, stable starting materials (trifluoroethylimidoyl chloride) and achieves significantly higher yields (up to 98%) under milder conditions without high-pressure CO gas.
Q: Can this synthetic route be applied to complex drug molecules like Rutaecarpine?
A: Yes, the patent explicitly demonstrates the successful application of this methodology in the efficient total synthesis of the bioactive alkaloid Rutaecarpine, achieving a robust 77% overall yield across three steps, proving its viability for complex pharmaceutical manufacturing.
Q: Is the use of toxic carbon monoxide gas required for this carbonylation reaction?
A: No, the process eliminates the need for hazardous high-pressure carbon monoxide gas cylinders by employing TFBen (1,3,5-tricarboxylic acid phenol ester) as a solid, safe, and easy-to-handle carbon monoxide surrogate that releases CO in situ under heating.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 2-Trifluoromethyl Quinazolinone Supplier
At NINGBO INNO PHARMCHEM, we recognize the transformative potential of this palladium-catalyzed carbonylation technology for the production of high-purity pharmaceutical intermediates. As a dedicated CDMO partner, we possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the transition from laboratory bench to industrial reactor is seamless and efficient. Our state-of-the-art facilities are equipped with rigorous QC labs capable of verifying stringent purity specifications, guaranteeing that every batch of 2-trifluoromethyl quinazolinone or related derivative meets the exacting standards required for clinical and commercial drug manufacturing.
We invite you to collaborate with us to leverage this innovative synthetic route for your specific project needs. By engaging with our technical procurement team, you can request a Customized Cost-Saving Analysis tailored to your target molecule, along with specific COA data and route feasibility assessments. Whether you require small quantities for early-stage research or bulk supply for commercial launch, our commitment to quality, safety, and cost-effectiveness makes us the ideal partner for your quinazolinone supply chain requirements.