Advanced Synthesis of N-N Axis Chiral Pyrrole Derivatives for Pharmaceutical Applications
The pharmaceutical industry is constantly seeking novel scaffolds that offer unique biological profiles, particularly in the realm of oncology. Patent CN114524701B introduces a groundbreaking methodology for the synthesis of N-N axis chiral pyrrole derivatives, a class of compounds that has historically been difficult to access with high stereochemical purity. These derivatives are not merely structural curiosities; they have demonstrated potent cytotoxic activity against QGP-1 pancreatic tumor cells, marking them as high-value targets for drug discovery programs. The core innovation lies in the utilization of chiral phosphoric acid catalysts to drive an asymmetric condensation reaction between indoleamines or pyrroleamines and 1,4-diketone derivatives. This approach bypasses the limitations of traditional dynamic kinetic resolution, enabling the direct construction of the chiral N-N axis with exceptional enantioselectivity. As depicted in the general structural formulas below, the versatility of this chemistry allows for extensive diversification of the R-groups, facilitating the rapid generation of compound libraries for SAR studies.
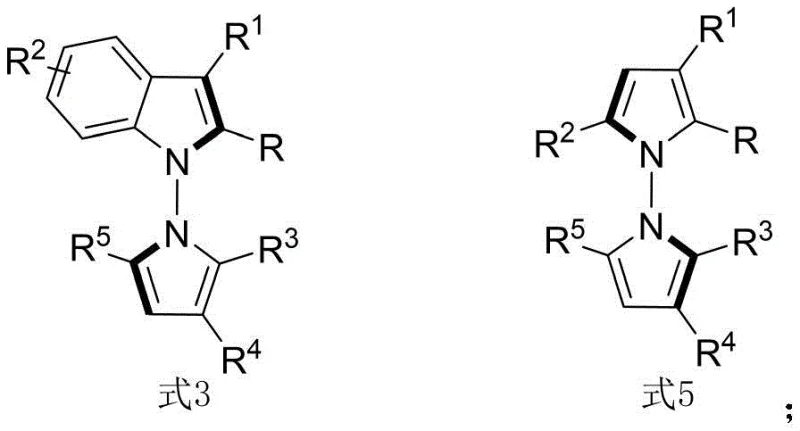
For R&D directors evaluating new synthetic routes, the ability to access complex chiral architectures efficiently is paramount. The traditional landscape for synthesizing N-N axis chiral compounds has been fraught with challenges, often relying on multi-step sequences or resolution processes that inherently cap the maximum theoretical yield at 50%. Furthermore, existing methods frequently necessitate harsh reaction conditions or expensive transition metal catalysts that introduce contamination risks requiring rigorous downstream purification. In contrast, the novel approach detailed in this patent leverages organocatalysis to achieve what was previously unattainable: a one-pot, in-situ ring formation strategy that constructs the pyrrole core and establishes the chiral axis simultaneously. This method operates under remarkably mild conditions, typically at room temperature (25°C), which preserves sensitive functional groups and minimizes energy consumption. The reaction scope is exceptionally broad, accommodating a wide array of substituents including halogens, alkoxy groups, and various aryl moieties without compromising stereoselectivity.
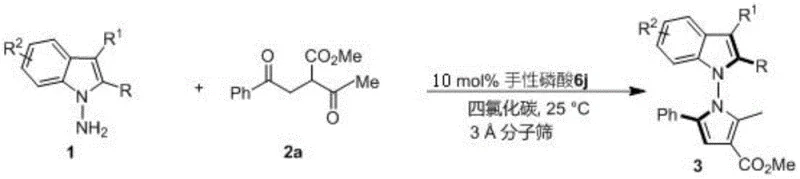
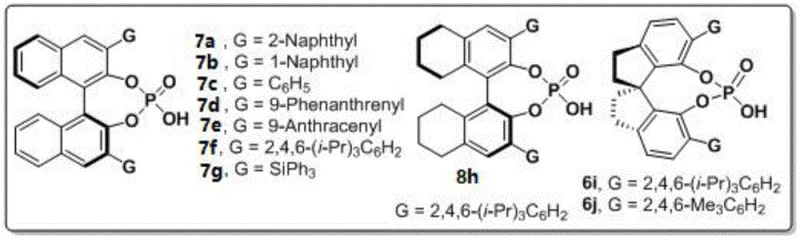
The mechanistic elegance of this transformation is driven by the precise spatial arrangement provided by the chiral phosphoric acid catalyst. Specifically, the patent highlights the superiority of spiro-binaphthyl backbone derivatives, such as catalyst 6j, which features a 2,4,6-trimethylphenyl group at the 3,3'-position. This bulky substituent creates a well-defined chiral pocket that effectively discriminates between the prochiral faces of the reacting species through a network of hydrogen bonding interactions. The catalyst activates the 1,4-diketone substrate while simultaneously orienting the amine nucleophile, ensuring that the cyclization proceeds through a single favored transition state. This results in products with enantiomeric excess (ee) values reaching as high as 96%, a level of purity that significantly reduces the burden on downstream chiral separation processes. Moreover, the mechanism inherently suppresses the formation of regioisomers and other side products, leading to cleaner reaction profiles and simplified purification workflows.
To facilitate the adoption of this technology in your laboratory, we have outlined the generalized operational parameters derived from the patent examples. The synthesis is robust and forgiving, utilizing standard laboratory equipment and reagents that are readily available from commercial suppliers. The process begins with the preparation of the reaction mixture in carbon tetrachloride, a solvent chosen for its ability to support the specific hydrogen-bonding network required for high induction. The addition of 3A molecular sieves plays a critical role in sequestering water generated during the condensation, thereby driving the equilibrium towards product formation. Detailed standardized synthesis steps follow below, providing a clear roadmap for replicating these high-value intermediates.
From a procurement and supply chain perspective, this synthetic route offers compelling advantages that translate directly into cost efficiency and operational reliability. The elimination of transition metals is a significant economic driver; it removes the need for expensive metal salts and, more importantly, obviates the costly and time-consuming heavy metal scavenging steps often required to meet regulatory limits for pharmaceutical ingredients. The use of organocatalysts like chiral phosphoric acids, while specialized, allows for lower loadings (typically 10 mol%) and easier removal compared to metal complexes. Furthermore, the reaction conditions are inherently safe and scalable. Operating at ambient temperature (25°C) eliminates the need for energy-intensive heating or cryogenic cooling systems, reducing the facility's utility costs and carbon footprint. The simplicity of the workup—filtration followed by concentration and standard column chromatography—ensures that the process can be transferred from gram-scale discovery to kilogram-scale production with minimal re-optimization.
- Cost Reduction in Manufacturing: The organocatalytic nature of this process fundamentally alters the cost structure of producing chiral pyrroles. By avoiding precious metals such as palladium or rhodium, the raw material costs are stabilized against volatile market fluctuations. Additionally, the high atom economy and excellent yields (often exceeding 90%) mean that less starting material is wasted, directly improving the cost-per-kilogram of the final API intermediate. The streamlined purification process further reduces solvent consumption and labor hours associated with complex workups.
- Enhanced Supply Chain Reliability: The starting materials, including various substituted indoleamines and 1,4-diketones, are structurally simple and can be sourced from multiple global suppliers, mitigating the risk of single-source dependency. The robustness of the reaction against minor variations in conditions ensures consistent batch-to-batch quality, which is critical for maintaining uninterrupted supply to downstream drug manufacturers. The mild conditions also reduce wear and tear on reactor vessels, extending equipment lifespan and reducing maintenance downtime.
- Scalability and Environmental Compliance: Scaling this reaction is straightforward due to the absence of exothermic hazards associated with strong bases or oxidants. The use of carbon tetrachloride, while requiring careful handling, is a well-understood solvent in industrial settings with established recovery and recycling protocols. The overall process generates minimal hazardous waste compared to traditional multi-step syntheses, aligning with modern green chemistry principles and simplifying environmental compliance reporting for large-scale facilities.
Understanding the technical nuances of this synthesis is crucial for successful implementation. We have compiled a set of frequently asked questions that address common concerns regarding catalyst selection, substrate scope, and biological evaluation. These insights are drawn directly from the experimental data provided in the patent, offering a realistic view of what can be achieved with this methodology. Whether you are concerned about the stability of specific functional groups or the reproducibility of the enantioselectivity, the following section provides the clarity needed to make informed decisions about integrating this technology into your pipeline.
- Prepare the reaction mixture by combining indoleamine or pyrroleamine substrates with 1,4-diketone derivatives in carbon tetrachloride solvent.
- Add 3A molecular sieves as an additive and introduce the chiral phosphoric acid catalyst (preferably catalyst 6j) at a loading of 10 mol%.
- Stir the reaction at room temperature (25°C) until completion monitored by TLC, then filter, concentrate, and purify via silica gel column chromatography.
Frequently Asked Questions (FAQ)
Q: What is the primary advantage of this synthesis method over conventional routes?
A: This method utilizes an in-situ ring formation strategy catalyzed by chiral phosphoric acids, offering extremely high enantioselectivity (up to 96% ee) and high yields under mild room temperature conditions, unlike traditional methods which often require harsh conditions or dynamic kinetic resolution.
Q: What biological activity do these N-N axis chiral pyrrole derivatives exhibit?
A: Biological testing indicates that these derivatives possess strong cytotoxic activity against QGP-1 pancreatic tumor cells, with specific compounds showing high sensitivity and low IC50 values, suggesting significant potential for anticancer drug development.
Q: Is this process suitable for large-scale industrial production?
A: Yes, the process is highly suitable for scale-up due to its mild reaction conditions (25°C), use of conventional solvents like carbon tetrachloride, simple workup procedures involving filtration and concentration, and the availability of diverse substrates.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable N-N Axis Chiral Pyrrole Derivative Supplier
The synthesis of N-N axis chiral pyrrole derivatives represents a significant leap forward in the construction of complex heterocyclic scaffolds for medicinal chemistry. At NINGBO INNO PHARMCHEM, we recognize the potential of this technology to accelerate the development of next-generation anticancer therapeutics. As a premier CDMO partner, we possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production. Our state-of-the-art facilities are equipped to handle the specific requirements of organocatalytic processes, ensuring that stringent purity specifications are met consistently. Our rigorous QC labs utilize advanced chiral HPLC and NMR spectroscopy to verify the enantiomeric excess and structural integrity of every batch, guaranteeing that the material you receive is ready for immediate use in biological assays or further synthetic elaboration.
We invite you to explore the possibilities of this innovative chemistry for your specific drug discovery projects. Our technical team is prepared to conduct a Customized Cost-Saving Analysis tailored to your target molecule, identifying opportunities to optimize the synthetic route for maximum efficiency. Please contact our technical procurement team today to request specific COA data for our catalog compounds or to discuss route feasibility assessments for your proprietary structures. Let us help you transform this promising academic research into a reliable, commercial reality.