Advanced Palladium-Catalyzed Synthesis of 2-Trifluoromethyl Imidazoles for Commercial Scale-Up
Advanced Palladium-Catalyzed Synthesis of 2-Trifluoromethyl Imidazoles for Commercial Scale-Up
The pharmaceutical and fine chemical industries are constantly seeking robust methodologies to access fluorinated heterocycles, particularly those containing the trifluoromethyl group, due to their profound impact on metabolic stability and lipophilicity. Patent CN111423381B introduces a groundbreaking preparation method for 2-trifluoromethyl substituted imidazole compounds, addressing critical bottlenecks in current synthetic routes. This technology leverages a transition metal palladium-catalyzed carbonylation series reaction, utilizing cheap and easily obtained starting materials such as trifluoroethylimidoyl chloride, propargylamine, and diaryl iodonium salts. The significance of this innovation lies in its ability to construct complex nitrogen-containing five-membered heterocycles under exceptionally mild conditions, specifically at 30°C, which contrasts sharply with the harsh thermal requirements of conventional cyclization protocols. As illustrated by the diverse bioactive scaffolds shown below, the utility of such imidazole cores extends far beyond simple intermediates, finding applications in high-value drugs and functional materials.
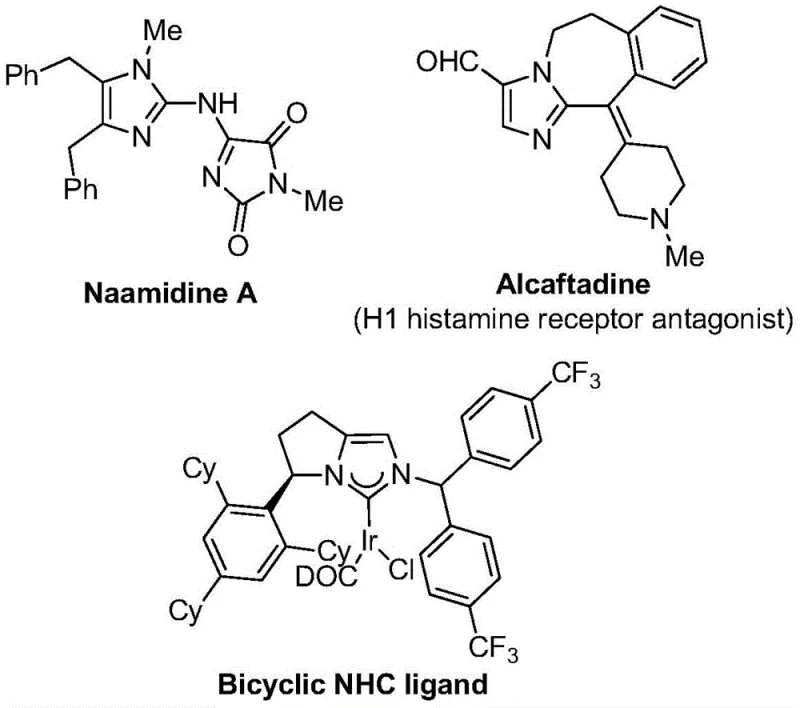
The strategic incorporation of the trifluoromethyl moiety into these heterocyclic backbones is not merely a structural modification but a critical design element for enhancing bioavailability and electronegativity in drug candidates. Traditional approaches often rely on direct trifluoromethylation using specialized synthons like trifluorodiazoethane, which can pose safety hazards and handling difficulties on a large scale. In contrast, the disclosed method employs trifluoroethylimidoyl chloride, a more stable and manageable reagent, thereby mitigating operational risks associated with volatile or explosive precursors. This shift in reagent strategy represents a pivotal advancement for process chemists aiming to develop safer, more sustainable manufacturing pathways for active pharmaceutical ingredients (APIs) and agrochemical intermediates.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of trifluoromethyl-substituted nitrogen-containing heterocycles has been plagued by significant technical challenges that hinder efficient commercial production. Conventional methods frequently necessitate the use of highly reactive and potentially dangerous trifluoromethyl synthons, such as trifluorodiazoethane, which require stringent safety controls and specialized equipment to manage explosion risks. Furthermore, many existing protocols demand elevated temperatures and prolonged reaction times, leading to increased energy consumption and the formation of complex impurity profiles that are difficult to separate. The reliance on harsh conditions often limits the scope of compatible functional groups, forcing chemists to employ tedious protection and deprotection strategies that add unnecessary steps and reduce overall atom economy. Additionally, the direct introduction of the CF3 group onto pre-formed heterocycles can suffer from poor regioselectivity, resulting in mixtures of isomers that compromise the purity required for pharmaceutical applications.
The Novel Approach
The novel approach detailed in patent CN111423381B circumvents these limitations through an elegant multicomponent coupling strategy driven by palladium catalysis. By utilizing trifluoroethylimidoyl chloride, propargylamine, and diaryl iodonium salts as the foundational building blocks, this method constructs the imidazole ring and installs the trifluoromethyl group simultaneously in a single pot. The reaction proceeds efficiently at a remarkably low temperature of 30°C, drastically reducing the thermal load on the process and minimizing thermal degradation of sensitive substrates. The use of diaryl iodonium salts as arylating agents provides a highly reactive yet controllable source of the aryl group, enabling excellent yields even with sterically hindered or electronically diverse substrates. This convergent synthesis not only streamlines the workflow by reducing the number of isolation steps but also enhances the overall sustainability of the process by avoiding the generation of excessive waste associated with multi-step linear syntheses.
Mechanistic Insights into Palladium-Catalyzed Carbonylation Cyclization
The mechanistic pathway of this transformation is a sophisticated cascade of organometallic events that ensures high fidelity in product formation. The cycle initiates with the formation of a trifluoroacetamidine intermediate via an intermolecular carbon-nitrogen bond promotion facilitated by the alkaline environment provided by sodium bicarbonate. Following isomerization, the palladium catalyst, generated in situ from PdCl2 and PPh3, engages with the alkyne moiety of the propargylamine derivative through a palladation event, yielding a key alkenyl palladium intermediate. This species undergoes a subsequent isomerization to form a more stable alkyl palladium intermediate, setting the stage for the crucial carbonylation step. Uniquely, this process avoids the use of external carbon monoxide gas cylinders; instead, it utilizes a formic acid and acetic anhydride mixture to release CO in situ, which inserts into the palladium-carbon bond to generate an acyl palladium species.
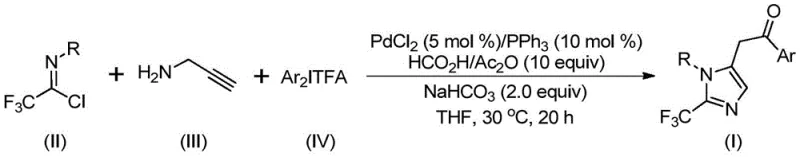
The final stages of the catalytic cycle involve the oxidative addition of the diaryl iodonium salt to the acyl palladium intermediate, forming a high-valent tetravalent palladium complex. This step is critical for introducing the aryl substituent at the 5-position of the imidazole ring with high precision. The cycle concludes with a reductive elimination step that releases the final 2-trifluoromethyl-substituted imidazole product and regenerates the active palladium(0) catalyst for the next turnover. This intricate mechanism highlights the dual role of the palladium center in mediating both the carbonylation and the arylation events, ensuring that the reaction proceeds with high atom economy and minimal side reactions. The compatibility of this mechanism with a wide range of electronic environments on the aryl rings (R and Ar groups) underscores the robustness of the catalytic system, making it suitable for synthesizing a diverse library of analogs for structure-activity relationship (SAR) studies.
How to Synthesize 2-Trifluoromethyl Imidazoles Efficiently
Implementing this synthesis in a laboratory or pilot plant setting requires careful attention to reagent stoichiometry and reaction parameters to maximize yield and purity. The protocol specifies a molar ratio of palladium chloride to triphenylphosphine to sodium bicarbonate of approximately 0.05:0.1:2, ensuring sufficient catalytic activity and neutralization of acidic byproducts. The reaction is typically conducted in tetrahydrofuran (THF), which has been identified as the optimal solvent for dissolving all components and promoting the reaction kinetics effectively. While the standard reaction time is cited between 16 to 24 hours, monitoring conversion via TLC or HPLC is recommended to determine the precise endpoint for specific substrate combinations. The detailed standardized synthesis steps see the guide below.
- Combine palladium chloride (5 mol%), triphenylphosphine (10 mol%), sodium bicarbonate, acetic anhydride, and formic acid in an organic solvent such as THF.
- Add trifluoroethylimidoyl chloride, propargylamine, and diaryl iodonium salt to the reaction mixture under stirring.
- Maintain the reaction at 30°C for 18 to 20 hours, then filter and purify via column chromatography to isolate the target imidazole.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this patented methodology offers tangible strategic benefits that extend beyond mere technical feasibility. The primary advantage lies in the utilization of commodity chemicals as starting materials; propargylamine, aromatic amines (precursors to imidoyl chlorides), and aryl boronic acids (precursors to iodonium salts) are widely available in the global chemical market, ensuring a stable and resilient supply chain. This reliance on bulk chemicals mitigates the risk of supply disruptions often associated with exotic or custom-synthesized reagents, thereby enhancing the continuity of production schedules for critical intermediates. Furthermore, the mild reaction conditions translate directly into operational cost efficiencies, as the process does not require specialized high-pressure reactors or extensive heating infrastructure, allowing for deployment in standard glass-lined or stainless steel vessels.
- Cost Reduction in Manufacturing: The economic viability of this process is significantly enhanced by the elimination of expensive and hazardous reagents typically used in trifluoromethylation chemistry. By replacing high-pressure carbon monoxide gas with a safe formic acid/acetic anhydride mixture, the facility saves on the capital expenditure required for gas handling systems and safety certifications. Additionally, the high reaction efficiency and substrate compatibility mean that fewer purification cycles are needed to achieve pharmaceutical-grade purity, reducing solvent consumption and waste disposal costs. The use of relatively inexpensive palladium chloride as the catalyst precursor, combined with the ability to potentially recover and recycle the precious metal, further drives down the cost of goods sold (COGS) for the final API intermediate.
- Enhanced Supply Chain Reliability: The robustness of the reaction against varying electronic properties of the substrates ensures that supply chains remain agile even when specific raw material grades fluctuate. Since the method tolerates halogens, nitro groups, and alkyl substituents without significant loss in yield, manufacturers can source alternative suppliers for starting materials without needing to re-validate the entire process. This flexibility is crucial for maintaining long-term supply contracts with multinational pharmaceutical clients who demand consistent quality and uninterrupted delivery. The scalability of the reaction from gram to kilogram levels, as demonstrated in the patent examples, provides a clear pathway for rapid scale-up to meet surging market demand without the need for extensive process re-engineering.
- Scalability and Environmental Compliance: From an environmental, health, and safety (EHS) perspective, this process aligns well with modern green chemistry principles. The avoidance of toxic CO gas and the operation at near-ambient temperatures reduce the facility's carbon footprint and energy usage. The post-treatment procedure involves straightforward filtration and column chromatography, which are well-understood unit operations that generate manageable waste streams. This simplifies the regulatory compliance burden associated with hazardous waste disposal and worker safety, making the technology attractive for manufacturing in regions with strict environmental regulations. The ability to produce high-purity products with minimal byproducts also reduces the load on wastewater treatment plants, contributing to a more sustainable manufacturing ecosystem.
Frequently Asked Questions (FAQ)
The following questions address common technical inquiries regarding the implementation and optimization of this synthesis route, derived directly from the experimental data and specifications within the patent documentation. Understanding these nuances is essential for process development teams aiming to integrate this technology into their existing portfolios. The answers provided reflect the specific conditions and outcomes observed during the validation of this method, ensuring accuracy and reliability for decision-making purposes.
Q: What is the role of formic acid and acetic anhydride in this synthesis?
A: In this patented process, the mixture of formic acid and acetic anhydride serves as an efficient carbon monoxide (CO) surrogate. Instead of using hazardous high-pressure CO gas, this system generates CO in situ under mild conditions (30°C), facilitating the carbonylation step of the palladium catalytic cycle safely and effectively.
Q: Can this method tolerate diverse functional groups on the aryl rings?
A: Yes, the methodology demonstrates excellent substrate compatibility. The patent data confirms that various substituents including methyl, tert-butyl, methoxy, chloro, bromo, trifluoromethyl, and nitro groups on both the imidoyl chloride and the diaryl iodonium salt are well-tolerated, yielding products with high efficiency (up to 97% yield in optimized cases).
Q: Is the reaction scalable for industrial production?
A: The process is designed for scalability. It utilizes commercially available starting materials like propargylamine and operates at a low temperature of 30°C, which significantly reduces energy consumption compared to traditional high-temperature heterocycle syntheses. The simple workup involving filtration and chromatography further supports its potential for large-scale manufacturing.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 2-Trifluoromethyl Imidazole Supplier
At NINGBO INNO PHARMCHEM, we recognize the transformative potential of this palladium-catalyzed technology in accelerating the development of next-generation therapeutics. Our team of expert process chemists possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your transition from benchtop discovery to full-scale manufacturing is seamless and efficient. We are committed to delivering high-purity 2-trifluoromethyl imidazole derivatives that meet stringent purity specifications, supported by our rigorous QC labs equipped with state-of-the-art analytical instrumentation. Our dedication to quality assurance guarantees that every batch supplied adheres to the highest industry standards, providing you with the confidence needed to advance your clinical programs.
We invite you to collaborate with us to leverage this innovative synthetic route for your specific project needs. By partnering with our technical procurement team, you can request a Customized Cost-Saving Analysis tailored to your volume requirements and target specifications. We encourage you to contact us today to obtain specific COA data and route feasibility assessments, allowing us to demonstrate how our advanced manufacturing capabilities can optimize your supply chain and reduce your overall development timelines.