Advanced Rhodium-Catalyzed Synthesis of Quinazolinone Derivatives for Commercial Scale-Up
Advanced Rhodium-Catalyzed Synthesis of Quinazolinone Derivatives for Commercial Scale-Up
The pharmaceutical industry continuously seeks efficient pathways to access complex polycyclic nitrogen-containing heterocycles, particularly those embodying the isoindolo[1,2-b]quinazolin-10(12H)-one skeleton found in bioactive natural products like Luotonin A, B, and E, as well as anticancer agents such as Belotecan (CKD-602). A significant breakthrough in this domain is detailed in Chinese Patent CN111592549B, which discloses a novel preparation method for quinazolinone derivatives via a rhodium-catalyzed [4+1] cyclization. This technology leverages continuous positioning group-assisted ortho C-H bond functionalization coupled with a previously unreported rhodium-catalyzed carbonyl alpha-amination process. By utilizing simple and readily available reagents to construct the four-ring core in a single step, this methodology addresses critical bottlenecks in the synthesis of high-value pharmaceutical intermediates. The ability to generate diverse aroylated luotonin A derivatives through this streamlined route offers substantial implications for reducing lead times in drug discovery and optimizing manufacturing costs for active pharmaceutical ingredients.
![Chemical structures of bioactive isoindolo[1,2-b]quinazolin-10(12H)-one derivatives including Luotonin A, B, E, 14-azacamptothecin, and Belotecan](/insights/img/quinazolinone-rhodium-catalysis-pharma-supplier-20260304215228-01.webp)
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the construction of the tetracyclic quinazolinone skeleton has been a formidable challenge in organic synthesis, often necessitating convoluted multi-step sequences that hinder commercial viability. Prior art methods typically rely on free radical domino cyclizations, which can suffer from poor selectivity and safety concerns due to the generation of reactive radical species. Alternatively, palladium-catalyzed approaches involving 2-bromo-N-(2-iodobenzyl)-benzamides require sequential cyanation, N-addition, and N-arylation steps, each introducing potential yield losses and purification complexities. Other strategies, such as the carbonylation of 2-bromobenzylamine with 2-bromoaniline or CuI/L-proline catalyzed Sonogashira couplings, depend heavily on expensive halogenated starting materials and precious metal catalysts that are difficult to remove to ppm levels required for GMP manufacturing. These legacy processes often result in extended production cycles, higher waste generation, and increased overall cost of goods sold (COGS), making them less attractive for large-scale supply chains.
The Novel Approach
In stark contrast, the methodology described in patent CN111592549B introduces a paradigm shift by employing a rhodium-catalyzed [4+1] cyclization between 2-arylquinazolin-4(3H)-ones and aroylthio ylides. This innovative strategy bypasses the need for pre-functionalized halogenated substrates, instead utilizing the inherent reactivity of the C-H bond adjacent to the directing group. The reaction proceeds through a sequential mechanism involving chelation-assisted ortho C-H functionalization followed by an intramolecular alpha-amination of the ketone moiety, a transformation never before achieved with rhodium catalysis. This one-pot ring-closing approach not only simplifies the synthetic tree but also enhances atom economy by incorporating the sulfur ylide as a C1 synthon directly into the heterocyclic framework. The versatility of this method is demonstrated by its compatibility with a wide range of substituents, allowing for the rapid generation of structural diversity essential for structure-activity relationship (SAR) studies in medicinal chemistry programs.
![General reaction scheme showing Rh-catalyzed [4+1] cyclization of 2-arylquinazolin-4(3H)-ones with aroylthio ylides to form quinazolinone derivatives](/insights/img/quinazolinone-rhodium-catalysis-pharma-supplier-20260304215228-02.webp)
Mechanistic Insights into Rhodium-Catalyzed C-H Functionalization and Alpha-Amination
The success of this transformation hinges on the unique ability of the pentamethylcyclopentadienyl rhodium(III) catalyst, specifically Cp*Rh(OAc)2·H2O, to activate inert C-H bonds under relatively mild thermal conditions. The mechanism initiates with the coordination of the rhodium species to the nitrogen atom of the quinazolinone ring, acting as a directing group to facilitate ortho-metallation. This forms a stable five-membered rhodacycle intermediate, which subsequently reacts with the aroylthio ylide. Unlike traditional carbene insertions where sulfur ylides act as C2 synthons, in this specific [4+1] cyclization, the sulfur ylide functions as a C1 synthon. The critical and unprecedented step involves the nucleophilic attack of the nitrogen onto the carbonyl carbon of the ylide-derived intermediate, effecting an intramolecular alpha-amination. This step closes the fifth ring to form the isoindole unit fused to the quinazolinone core. The presence of silver salts (such as AgNO3) and copper additives (like Cu(OAc)2·H2O) plays a pivotal role in regenerating the active Rh(III) species and facilitating the elimination of the sulfur byproduct, ensuring the catalytic cycle continues efficiently without catalyst deactivation.
From an impurity control perspective, this mechanism offers distinct advantages over radical-based pathways. The concerted nature of the C-H activation and the specific coordination geometry imposed by the Cp*Rh ligand minimize the formation of regioisomers and polymeric byproducts often seen in non-directed functionalizations. Furthermore, the use of aroylthio ylides as stable solid reagents eliminates the handling hazards associated with diazo compounds or other unstable carbene precursors. The reaction conditions, optimized at 120°C in 1,2-dichloroethane (DCE), provide a balance between kinetic energy for C-H cleavage and thermal stability of the sensitive heterocyclic products. This precise control over the reaction trajectory ensures a cleaner crude profile, which significantly reduces the burden on downstream purification processes such as column chromatography or recrystallization, thereby enhancing the overall purity of the final pharmaceutical intermediate.
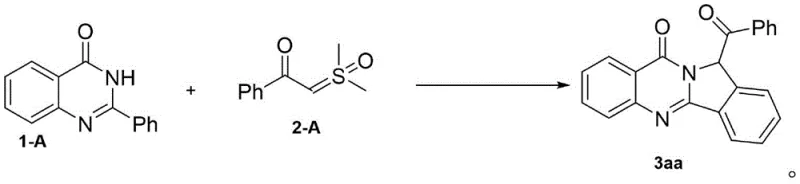
How to Synthesize 12-Benzoylisoindolo[1,2-b]quinazolin-10(12H)-ones Efficiently
To implement this cutting-edge synthesis in a laboratory or pilot plant setting, operators must adhere to the optimized protocol established in the patent examples. The process begins with the precise weighing of the 2-arylquinazolin-4(3H)-one substrate and the aroylthio ylide coupling partner, typically in a molar ratio of 1:1.5 to ensure complete consumption of the limiting reagent. The catalyst system, comprising Cp*Rh(OAc)2·H2O (5 mol%), AgNO3 (50 mol%), and Cu(OAc)2·H2O (0.5 to 2 equivalents), is added to a Schlenk tube equipped with a magnetic stir bar. Following the addition of the organic solvent, preferably 1,2-dichloroethane (DCE), the reaction vessel is subjected to multiple vacuum-nitrogen purge cycles to establish a strictly inert atmosphere, which is crucial for preventing catalyst oxidation. The detailed standardized synthesis steps, including specific workup procedures and purification parameters, are outlined in the guide below.
- Charge a Schlenk tube with 2-arylquinazolin-4(3H)-one, aroylthio ylide, Cp*Rh(OAc)2·H2O catalyst, AgNO3 promoter, and Cu(OAc)2·H2O additive.
- Add 1,2-dichloroethane (DCE) as the solvent and purge the system with nitrogen gas three times to ensure an inert atmosphere.
- Heat the reaction mixture to 120°C for 12 hours, then cool, filter, concentrate, and purify via column chromatography to isolate the target product.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this rhodium-catalyzed methodology presents a compelling value proposition centered on cost efficiency and supply reliability. By shifting away from multi-step halogenation and cross-coupling sequences, manufacturers can drastically reduce the number of unit operations required to produce the core quinazolinone scaffold. This consolidation of steps translates directly into lower labor costs, reduced solvent consumption, and minimized waste disposal fees, all of which contribute to a more competitive pricing structure for the final intermediate. Furthermore, the reliance on commercially available 2-arylquinazolinones and sulfur ylides mitigates the risk of supply chain disruptions often associated with custom-synthesized halogenated building blocks, ensuring a more stable and predictable raw material flow for long-term production campaigns.
- Cost Reduction in Manufacturing: The elimination of expensive palladium catalysts and the reduction in synthetic steps significantly lower the direct material costs. Additionally, the high atom economy of the [4+1] cyclization minimizes waste generation, leading to substantial cost savings in environmental compliance and waste treatment. The use of robust base metal additives like copper further optimizes the catalyst cost profile compared to systems relying solely on precious metals.
- Enhanced Supply Chain Reliability: The starting materials for this process are structurally simple and can be sourced from multiple global suppliers, reducing dependency on single-source vendors. The robustness of the reaction conditions allows for flexible scheduling and batch sizing, enabling manufacturers to respond quickly to fluctuating demand from downstream API producers without compromising on quality or delivery timelines.
- Scalability and Environmental Compliance: The protocol operates in standard organic solvents and at moderate temperatures, making it highly amenable to scale-up from gram to kilogram scales using existing reactor infrastructure. The absence of hazardous radical initiators or toxic gaseous reagents simplifies the safety assessment and regulatory approval process, facilitating faster technology transfer to commercial manufacturing sites while adhering to strict environmental, health, and safety (EHS) standards.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this novel quinazolinone synthesis technology. These insights are derived directly from the experimental data and beneficial effects reported in the patent documentation, providing clarity on the practical application of the method for industrial partners seeking to optimize their synthetic routes.
Q: What represents the primary innovation in this quinazolinone synthesis method?
A: The primary innovation lies in the first reported rhodium-catalyzed intramolecular α-amination of ketones combined with sequential chelation-assisted ortho C–H functionalization, enabling a direct [4+1] cyclization that was previously unachievable with standard palladium or copper methods.
Q: How does this method improve upon conventional synthetic routes for Luotonin A analogs?
A: Conventional routes often require multi-step sequences involving radical domino cyclizations or expensive palladium-catalyzed carbonylations with halogenated precursors. This new method utilizes readily available 2-arylquinazolin-4(3H)-ones and sulfur ylides to form the core ring structure in a single operational step, significantly simplifying the process.
Q: Is this synthesis strategy suitable for large-scale industrial production?
A: Yes, the protocol employs robust reaction conditions (100-140°C) and commercially available catalysts (Cp*Rh(OAc)2·H2O) in standard organic solvents like DCE. The tolerance for various substituents and the high isolated yields (up to 78%) indicate strong potential for commercial scale-up.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Quinazolinone Derivatives Supplier
At NINGBO INNO PHARMCHEM, we recognize the transformative potential of advanced C-H functionalization technologies in modern drug development. As a premier CDMO partner, we possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that innovative academic discoveries like the rhodium-catalyzed [4+1] cyclization are successfully translated into robust industrial processes. Our state-of-the-art facilities are equipped with rigorous QC labs capable of meeting stringent purity specifications required for pharmaceutical intermediates, guaranteeing that every batch of quinazolinone derivative delivered meets the highest quality standards for your critical applications.
We invite you to collaborate with our technical team to explore how this efficient synthesis route can enhance your project economics. By requesting a Customized Cost-Saving Analysis, you can gain a deeper understanding of the specific financial benefits applicable to your portfolio. We encourage you to contact our technical procurement team today to obtain specific COA data and comprehensive route feasibility assessments tailored to your unique manufacturing requirements.