Advanced FeCl3-Catalyzed Synthesis of 5-Trifluoromethyl-1,2,4-Triazole Derivatives for Commercial Scale-Up
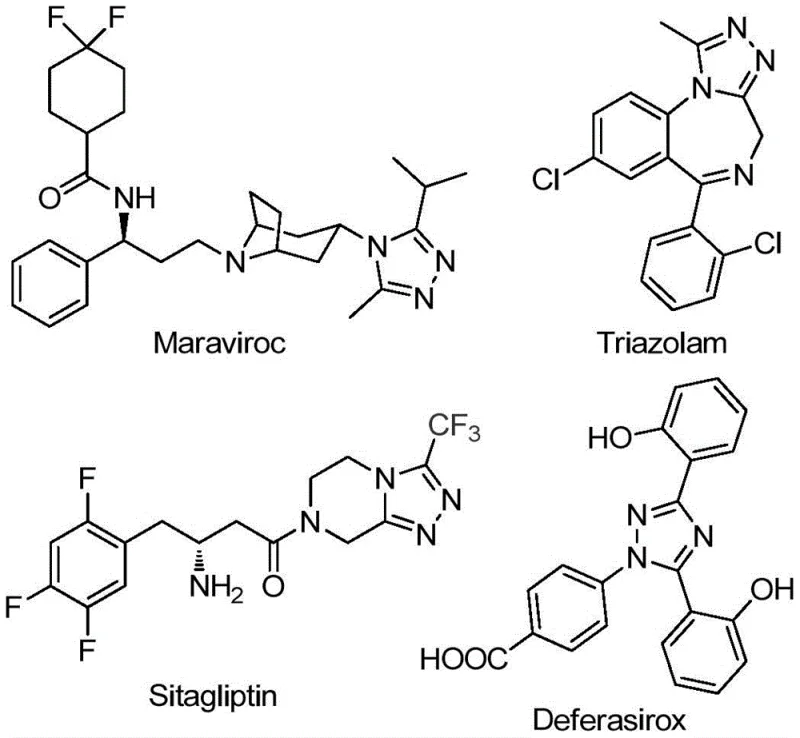
The pharmaceutical and agrochemical industries continuously seek robust synthetic routes for nitrogen-containing heterocycles, particularly 1,2,4-triazole derivatives, due to their prevalence in bioactive molecules. Patent CN111978265B discloses a groundbreaking preparation method for 5-trifluoromethyl substituted 1,2,4-triazole derivatives that addresses long-standing challenges in efficiency and operational simplicity. The introduction of a trifluoromethyl group into these heterocyclic frameworks is strategically vital, as it significantly enhances electronegativity, metabolic stability, and lipophilicity, properties essential for modern drug design. As illustrated in the structural diversity of marketed drugs like Maraviroc, Triazolam, Sitagliptin, and Deferasirox, the 1,2,4-triazole core is a privileged scaffold in medicinal chemistry.
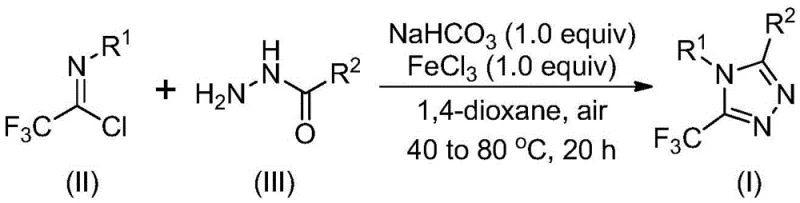
This novel methodology leverages a ferric chloride-promoted cyclization strategy that operates under remarkably mild conditions, avoiding the stringent anhydrous and oxygen-free environments typically demanded by conventional heterocycle synthesis. For R&D directors and process chemists, this represents a paradigm shift towards more forgiving and scalable chemistry. The ability to utilize cheap and readily available starting materials, specifically acyl hydrazides and trifluoroethylimidoyl chlorides, positions this technology as a highly attractive option for cost reduction in pharmaceutical intermediate manufacturing. Furthermore, the process demonstrates exceptional functional group tolerance, allowing for the synthesis of complex derivatives without extensive protecting group strategies, thereby streamlining the overall synthetic timeline.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of trifluoromethyl-substituted 1,2,4-triazoles has been plagued by significant operational hurdles that impede efficient commercial production. Traditional literature methods often rely on the condensation of 3,5-ditrifluoromethyl-1,3,4-oxadiazoles with primary amines or the cyclization of trifluoromethyl hydrazides with amidines. These legacy routes are frequently characterized by harsh reaction conditions, requiring extreme temperatures or pressures that pose safety risks and increase energy consumption. Moreover, many established protocols suffer from narrow substrate scopes, failing to accommodate alkyl hydrazones effectively, which limits the chemical space accessible to medicinal chemists. The necessity for multi-step sequences and the generation of difficult-to-remove byproducts further exacerbate the cost and environmental burden, making these conventional methods less viable for large-scale supply chains focused on sustainability and efficiency.
The Novel Approach
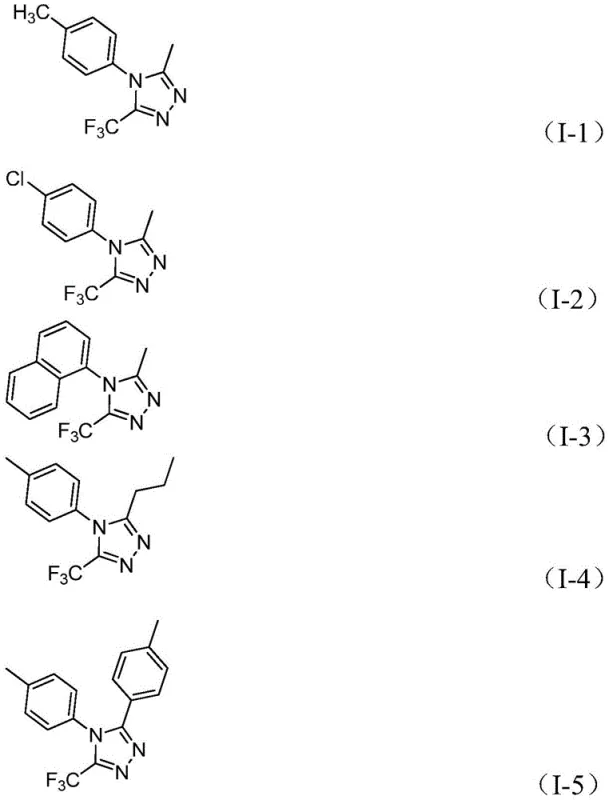
In stark contrast, the method disclosed in CN111978265B offers a streamlined, one-pot solution that dramatically simplifies the synthetic landscape. By employing a tandem reaction sequence initiated by base-promoted intermolecular carbon-nitrogen bond formation followed by Lewis acid-promoted intramolecular dehydration, this approach achieves high conversion rates with minimal waste. The versatility of this new route is evidenced by its ability to synthesize a wide array of derivatives, including those with diverse aryl and alkyl substitutions at the 3 and 4 positions of the triazole ring. As shown in the specific examples provided, the method successfully generates compounds like I-1 through I-5 with high purity and yield, demonstrating its practical utility for generating diverse chemical libraries.
The operational simplicity extends to the reaction conditions, which do not require inert atmosphere techniques, allowing the reaction to proceed in air. This feature alone drastically reduces the capital expenditure required for specialized reactor equipment and lowers the barrier for technology transfer from lab to plant. The use of 1,4-dioxane as a preferred solvent ensures good solubility for various substrates, facilitating homogeneous reaction kinetics that are crucial for consistent product quality. For procurement managers, this translates to a reliable pharmaceutical intermediate supplier capability, as the raw materials are commodity chemicals rather than exotic, hard-to-source reagents.
Mechanistic Insights into FeCl3-Catalyzed Cyclization
The core of this technological advancement lies in the dual-role catalytic system involving sodium bicarbonate and ferric chloride. The reaction mechanism is hypothesized to proceed through an initial base-promoted nucleophilic attack where the hydrazide nitrogen attacks the imidoyl chloride carbon, forming a trifluoroacetamidine intermediate. This step is critical for establishing the carbon-nitrogen framework necessary for the subsequent ring closure. Following this, the addition of the metal Lewis acid, specifically ferric chloride, activates the intermediate for intramolecular cyclization. The Lewis acidity of the iron center likely coordinates with the nitrogen atoms, increasing the electrophilicity of the adjacent carbon and facilitating the dehydration step that aromatizes the triazole ring. This mechanistic pathway avoids the high-energy barriers associated with thermal cyclization, allowing the reaction to proceed at moderate temperatures between 70°C and 90°C.
From an impurity control perspective, the mildness of the FeCl3-catalyzed system is a significant advantage. Harsh acidic or basic conditions often lead to the hydrolysis of sensitive functional groups or the formation of polymeric byproducts, complicating downstream purification. By maintaining a controlled pH environment with sodium bicarbonate and utilizing a specific Lewis acid promoter, the process minimizes side reactions such as over-alkylation or decomposition of the trifluoromethyl group. The result is a cleaner crude reaction profile, which reduces the load on purification units like column chromatography or crystallization steps. For quality assurance teams, this means a more consistent impurity profile and higher confidence in meeting stringent purity specifications required for GMP manufacturing of active pharmaceutical ingredients.
How to Synthesize 5-Trifluoromethyl-1,2,4-Triazole Derivatives Efficiently
The synthesis protocol outlined in the patent provides a clear roadmap for replicating these results in a laboratory or pilot plant setting. The procedure involves a sequential addition of reagents to manage the exothermic nature of the initial coupling and optimize the cyclization kinetics. Operators begin by dissolving the hydrazide and trifluoroethylimidoyl chloride in the organic solvent with sodium bicarbonate, stirring at a lower temperature range to ensure complete formation of the amidine intermediate before introducing the catalyst. Detailed standardized synthesis steps follow below.
- Mix sodium bicarbonate, trifluoroethylimide acid chloride, and hydrazide in an organic solvent like 1,4-dioxane.
- React the mixture at 30-50°C for 8-16 hours to form the intermediate trifluoroacetamidine derivatives.
- Add ferric chloride catalyst and heat to 70-90°C for 6-10 hours to complete the cyclization and dehydration.
Commercial Advantages for Procurement and Supply Chain Teams
For stakeholders focused on the bottom line and supply continuity, this patented process offers compelling economic and logistical benefits. The shift away from complex, multi-step syntheses towards a direct, catalytic cyclization fundamentally alters the cost structure of producing these valuable heterocycles. By eliminating the need for expensive transition metal catalysts like palladium or rhodium, which are common in cross-coupling approaches, the process relies on iron, one of the most abundant and inexpensive metals on earth. This substitution results in substantial cost savings on raw materials and removes the regulatory burden associated with heavy metal residue limits in final drug products.
- Cost Reduction in Manufacturing: The economic viability of this method is driven by the use of commodity-grade starting materials. Acyl chlorides and hydrazine hydrate, the precursors for the hydrazide component, are produced on a massive industrial scale, ensuring stable pricing and availability. Furthermore, the elimination of strict anhydrous conditions means that solvent drying and storage costs are significantly reduced. The process efficiency, characterized by high yields across a broad range of substrates, maximizes the throughput of existing reactor volumes, effectively lowering the cost per kilogram of the final intermediate without requiring new capital investment.
- Enhanced Supply Chain Reliability: Supply chain resilience is bolstered by the simplicity of the reagent list. Unlike specialized fluorinating agents or custom-synthesized heterocycles that may have single-source suppliers, the key inputs for this reaction are widely available from multiple global chemical vendors. This diversification mitigates the risk of supply disruptions caused by geopolitical issues or production failures at a single manufacturer. Additionally, the robustness of the reaction to air and moisture simplifies logistics, as raw materials do not require specialized shipping containers or cold chain management, further reducing transportation costs and lead times.
- Scalability and Environmental Compliance: Scaling chemical processes often introduces new safety and environmental challenges, but this methodology is inherently designed for expansion. The use of 1,4-dioxane, while requiring careful handling, is a well-understood solvent in the industry with established recovery and recycling protocols. The reaction generates minimal hazardous waste compared to older methods that might produce stoichiometric amounts of toxic byproducts. The ability to run the reaction at atmospheric pressure and moderate temperatures reduces the energy footprint of the manufacturing process, aligning with modern green chemistry principles and helping companies meet their sustainability goals while maintaining high production volumes.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis technology. These answers are derived directly from the experimental data and claims within the patent documentation, providing a factual basis for decision-making. Understanding these nuances is critical for project managers evaluating the feasibility of adopting this route for their specific pipeline candidates.
Q: What are the advantages of the FeCl3-catalyzed method over traditional triazole synthesis?
A: The FeCl3-catalyzed method described in patent CN111978265B eliminates the need for harsh anhydrous and oxygen-free conditions required by traditional methods. It utilizes cheap, readily available starting materials like hydrazides and trifluoroethylimide chlorides, significantly simplifying the operational complexity and reducing raw material costs compared to older routes involving oxadiazoles or complex nitrile cyclizations.
Q: Is this synthesis method suitable for large-scale industrial production?
A: Yes, the process is highly scalable. The patent explicitly states that the method can be easily expanded to the gram level and beyond, providing convenience for industrial scale production. The use of common solvents like 1,4-dioxane and inexpensive catalysts like ferric chloride ensures that the supply chain remains robust and cost-effective for commercial manufacturing.
Q: What is the substrate scope for R1 and R2 groups in this triazole synthesis?
A: The method demonstrates broad substrate tolerance. R1 can be substituted or unsubstituted aryl groups (e.g., phenyl with methyl, methoxy, or halogen substituents), while R2 accommodates alkyl, alkenyl, or aryl groups. This flexibility allows for the design and synthesis of diverse 3,4-disubstituted 1,2,4-triazole derivatives tailored for specific pharmaceutical applications.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 5-Trifluoromethyl-1,2,4-Triazole Derivative Supplier
At NINGBO INNO PHARMCHEM, we recognize the strategic value of efficient heterocycle synthesis in accelerating drug development timelines. Our team of expert process chemists has extensively evaluated the FeCl3-catalyzed route described in CN111978265B and confirmed its potential for robust commercial manufacturing. We possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your transition from benchtop discovery to clinical supply is seamless. Our state-of-the-art facilities are equipped with rigorous QC labs capable of meeting stringent purity specifications, guaranteeing that every batch of 5-trifluoromethyl-1,2,4-triazole derivative meets the highest international standards for pharmaceutical intermediates.
We invite you to collaborate with us to leverage this advanced technology for your next project. By partnering with our technical procurement team, you can access a Customized Cost-Saving Analysis tailored to your specific volume requirements and target molecules. We encourage you to contact us today to request specific COA data for our catalog compounds or to discuss route feasibility assessments for your proprietary candidates, ensuring a secure and cost-effective supply chain for your critical building blocks.