Scalable Metal-Free Synthesis of Trifluoromethyl 1,2,4-Triazine Compounds for Pharmaceutical Applications
Scalable Metal-Free Synthesis of Trifluoromethyl 1,2,4-Triazine Compounds for Pharmaceutical Applications
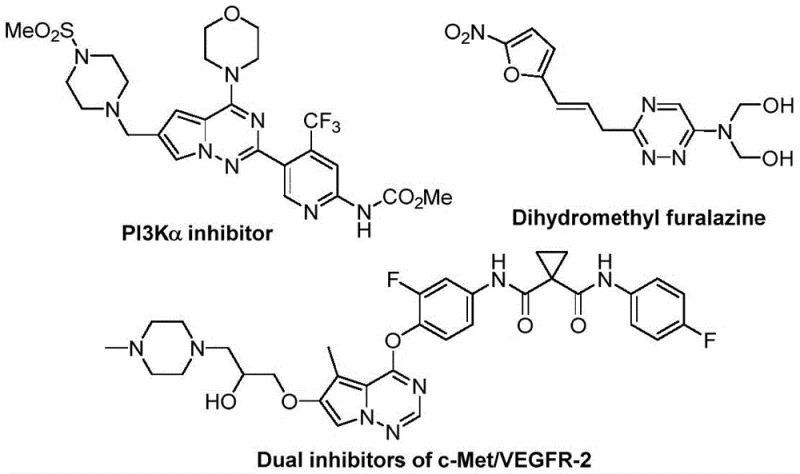
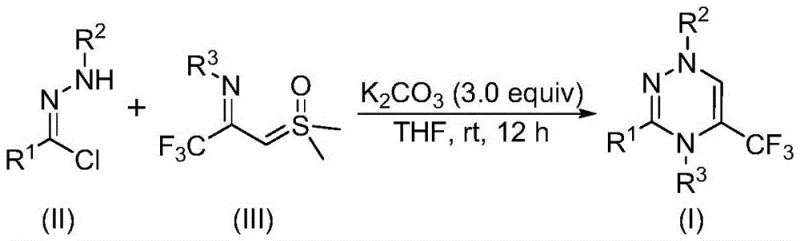
The pharmaceutical industry continuously seeks robust methodologies for constructing nitrogen-rich heterocyclic scaffolds, particularly those incorporating fluorine motifs which are pivotal for enhancing metabolic stability and bioavailability. Patent CN116253692A discloses a groundbreaking preparation method for trifluoromethyl-substituted 1,2,4-triazine compounds, addressing critical bottlenecks in current synthetic routes. This innovation leverages a synergistic [3+3] cycloaddition strategy between chlorohydrazones and trifluoroacetyl sulfur ylides, facilitated by potassium carbonate. The significance of this development cannot be overstated, as 1,2,4-triazine cores are ubiquitous in high-value therapeutic agents ranging from anticancer to antimalarial drugs. By enabling the efficient assembly of these complex architectures under ambient conditions, this technology offers a reliable pharmaceutical intermediate supplier pathway that aligns perfectly with modern green chemistry principles and industrial scalability requirements.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the construction of 1,2,4-triazine rings has relied heavily on condensation reactions between amidrazones and 1,2-diketones or alkynes, often necessitating harsh thermal conditions and extended reaction times. Traditional multicomponent reactions involving hydrazides and dicarbonyl compounds frequently suffer from poor atom economy and generate significant quantities of waste, complicating the purification process and driving up operational costs. Furthermore, many established protocols require the use of stoichiometric amounts of activating agents or toxic heavy metal catalysts, which pose severe challenges for regulatory compliance in API manufacturing due to strict limits on residual metals. The structural diversity achievable through these legacy methods is often restricted, limiting the ability of medicinal chemists to rapidly explore structure-activity relationships around the triazine core. Consequently, there is a persistent demand for more efficient, versatile, and environmentally benign synthetic strategies that can overcome these inherent limitations.
The Novel Approach
The methodology described in the patent represents a paradigm shift by utilizing readily available chlorohydrazones and trifluoroacetyl sulfur ylides as key building blocks in a streamlined one-pot transformation. This novel approach operates under remarkably mild conditions, typically between 20°C and 40°C, and crucially, proceeds efficiently under an air atmosphere without the need for rigorous exclusion of moisture or oxygen. The use of potassium carbonate as a benign, inexpensive base eliminates the reliance on strong, hazardous bases or expensive transition metal catalysts, thereby simplifying the workup procedure and reducing the environmental footprint. This metal-free protocol not only enhances the safety profile of the manufacturing process but also significantly broadens the scope of compatible functional groups, allowing for the synthesis of diverse derivatives with high structural fidelity. Such attributes make this method exceptionally attractive for the cost reduction in pharmaceutical intermediate manufacturing, offering a direct route to high-value targets with minimal processing overhead.
Mechanistic Insights into Potassium Carbonate-Promoted Cycloaddition
The reaction mechanism involves a sophisticated cascade initiated by the deprotonation of the chlorohydrazone substrate by potassium carbonate, leading to the elimination of hydrogen chloride and the generation of a reactive nitrile imine intermediate in situ. This highly electrophilic species then engages in a concerted [3+3] cycloaddition with the trifluoroacetyl sulfur ylide, a process that constructs the six-membered 1,2,4-triazine ring with excellent regioselectivity. The final step involves the elimination of dimethyl sulfoxide (DMSO) from the adduct, driving the equilibrium towards the formation of the aromatic triazine product. This mechanistic pathway is distinct from traditional cyclizations as it avoids high-energy intermediates and proceeds through a low-energy transition state facilitated by the unique electronic properties of the sulfur ylide. Understanding this mechanism is vital for process optimization, as it highlights the critical role of the base in generating the reactive dipole and the importance of the ylide structure in ensuring successful ring closure.
From an impurity control perspective, the cleanliness of this reaction is attributed to the high specificity of the [3+3] cycloaddition, which minimizes the formation of side products commonly associated with radical pathways or uncontrolled polymerization. The absence of transition metals precludes the formation of metal-complexed impurities, which are notoriously difficult to remove and often require specialized scavenging resins or recrystallization steps. Furthermore, the mild reaction conditions prevent the degradation of sensitive functional groups on the aromatic rings, ensuring that the final product retains the desired substitution pattern without unintended modifications. This high level of chemoselectivity translates directly into a simplified purification workflow, where standard column chromatography or crystallization is often sufficient to achieve pharmaceutical-grade purity. For R&D teams, this implies a faster turnaround time for generating compound libraries and a more predictable scale-up trajectory from milligram to kilogram scales.
How to Synthesize Trifluoromethyl 1,2,4-Triazine Efficiently
The practical execution of this synthesis is designed for ease of operation, requiring standard laboratory equipment and commercially available reagents that do not demand special storage or handling precautions. The process begins with the precise weighing of chlorohydrazone and trifluoroacetyl sulfur ylide, which are then suspended in an aprotic organic solvent such as tetrahydrofuran (THF) to ensure homogeneous mixing. Potassium carbonate is added as a solid promoter, and the mixture is stirred at room temperature for a duration of 10 to 14 hours, allowing the reaction to reach completion as monitored by TLC or HPLC. Upon completion, the reaction mixture is filtered to remove inorganic salts, and the filtrate is concentrated and subjected to silica gel chromatography to isolate the pure trifluoromethyl-substituted 1,2,4-triazine compound. Detailed standardized synthesis steps are provided in the guide below to ensure reproducibility and consistency across different batches.
- Combine potassium carbonate, chlorohydrazone, and trifluoroacetyl sulfur ylide in an organic solvent such as THF.
- Stir the reaction mixture at room temperature (20-40°C) under an air atmosphere for 10 to 14 hours.
- Filter the reaction mixture, mix with silica gel, and purify via column chromatography to isolate the final product.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this synthetic route offers transformative benefits that extend far beyond simple yield improvements, fundamentally altering the cost structure of producing these valuable heterocycles. The elimination of precious metal catalysts removes a significant variable cost component and mitigates the supply chain risks associated with the volatility of metal prices and availability. Additionally, the ability to run the reaction under air atmosphere reduces the capital expenditure required for specialized reactor infrastructure, such as nitrogen blanketing systems or gloveboxes, thereby lowering the barrier to entry for contract manufacturing organizations. The use of inexpensive inorganic bases like potassium carbonate further drives down raw material costs, while the mild temperature requirements result in substantial energy savings compared to processes requiring reflux or cryogenic cooling. These factors collectively contribute to a more resilient and cost-effective supply chain capable of meeting the demanding timelines of modern drug development programs.
- Cost Reduction in Manufacturing: The most immediate financial impact stems from the complete avoidance of expensive transition metal catalysts, which not only lowers the bill of materials but also drastically simplifies the downstream purification process. Without the need for metal scavengers or extensive washing protocols to meet strict residual metal specifications, the overall processing time is reduced, leading to lower labor and utility costs per kilogram of product. Furthermore, the high atom economy of the cycloaddition reaction ensures that a greater proportion of the starting materials are converted into the desired product, minimizing waste disposal fees and maximizing resource utilization. This economic efficiency makes the process highly competitive for large-scale production, offering significant margins for generic API manufacturers and custom synthesis providers alike.
- Enhanced Supply Chain Reliability: The reliance on commodity chemicals such as chlorohydrazones, sulfur ylides, and potassium carbonate ensures a stable and secure supply of raw materials, as these substances are produced by multiple global vendors with established distribution networks. Unlike specialized reagents that may have long lead times or single-source dependencies, the inputs for this reaction are readily available off-the-shelf, reducing the risk of production delays due to material shortages. The robustness of the reaction conditions also means that the process is less susceptible to variations in environmental factors, ensuring consistent output quality regardless of seasonal changes or facility locations. This reliability is crucial for maintaining continuous manufacturing operations and meeting the just-in-time delivery expectations of major pharmaceutical clients.
- Scalability and Environmental Compliance: Scaling this process from gram to tonnage levels is straightforward due to the absence of exothermic hazards or pressure buildup, allowing for safe operation in standard stainless steel reactors without the need for complex engineering controls. The generation of benign byproducts like DMSO and inorganic salts simplifies waste treatment and aligns with increasingly stringent environmental regulations regarding solvent emissions and heavy metal discharge. The green chemistry profile of this method enhances the sustainability credentials of the final product, which is becoming a key differentiator in supplier selection criteria for environmentally conscious pharmaceutical companies. Consequently, this technology supports the commercial scale-up of complex pharmaceutical intermediates while adhering to the highest standards of safety and environmental stewardship.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this novel synthesis method, providing clarity on its practical application and advantages over existing technologies. These insights are derived directly from the experimental data and comparative analysis presented in the patent documentation, ensuring accuracy and relevance for technical decision-makers. Understanding these nuances is essential for evaluating the feasibility of integrating this route into your current manufacturing portfolio or R&D pipeline.
Q: Does this synthesis require expensive transition metal catalysts?
A: No, the process utilizes inexpensive potassium carbonate as a promoter, completely eliminating the need for costly heavy metal catalysts and simplifying downstream purification.
Q: What are the reaction conditions regarding temperature and atmosphere?
A: The reaction proceeds efficiently at mild temperatures between 20°C and 40°C and does not require inert gas protection, operating successfully under standard air atmosphere.
Q: Is this method suitable for large-scale manufacturing?
A: Yes, the use of cheap, non-toxic reagents and mild conditions makes the process highly scalable and safe for commercial production without complex engineering controls.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Trifluoromethyl 1,2,4-Triazine Supplier
At NINGBO INNO PHARMCHEM, we recognize the strategic importance of accessing advanced synthetic technologies that drive innovation and efficiency in drug discovery and development. Our team of expert chemists possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that promising laboratory discoveries can be seamlessly translated into viable industrial processes. We are committed to delivering high-purity intermediates that meet stringent purity specifications, supported by our rigorous QC labs equipped with state-of-the-art analytical instrumentation. By leveraging our deep expertise in heterocyclic chemistry and process optimization, we can help you navigate the complexities of bringing trifluoromethyl-substituted triazines to market with speed and confidence.
We invite you to engage with our technical procurement team to discuss how this metal-free synthesis can be tailored to your specific project needs. Whether you require a Customized Cost-Saving Analysis for an existing program or need to evaluate the feasibility of a new target, we are ready to provide comprehensive support. Please contact us today to request specific COA data and route feasibility assessments, and let us partner with you to accelerate your journey from concept to commercial success.