Advanced Pd-Catalyzed Synthesis of Carbonyl-Bridged Biheterocyclic Compounds for Scalable Pharmaceutical Manufacturing
Introduction to Advanced Biheterocyclic Synthesis
The pharmaceutical and fine chemical industries are constantly seeking robust methodologies to construct complex heterocyclic scaffolds, which serve as the structural backbone for numerous bioactive molecules. Patent CN115353511A introduces a groundbreaking multicomponent strategy for the efficient synthesis of carbonyl-bridged biheterocyclic compounds, specifically targeting the fusion of indolinone and imidazole motifs. This technological advancement addresses critical bottlenecks in traditional heterocycle construction by leveraging a palladium-catalyzed carbonylation cascade that operates under exceptionally mild conditions. The significance of this invention lies not only in its chemical elegance but also in its practical applicability for generating high-value pharmaceutical intermediates. By integrating readily available starting materials such as trifluoroethylimidoyl chloride, propargylamine, and acrylamide derivatives, this process offers a streamlined pathway to structurally diverse targets that are otherwise difficult to access. For R&D directors and process chemists, this represents a pivotal shift towards more sustainable and operationally simple synthetic routes that maintain high purity standards while minimizing hazardous waste generation.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the construction of biheterocyclic systems containing both indolinone and imidazole rings has relied on laborious multi-step sequences or harsh reaction conditions that limit their utility in large-scale manufacturing. Traditional approaches often involve the direct coupling of two pre-formed heterocyclic substrates, which frequently suffers from low atom economy and poor regioselectivity, leading to complex purification challenges and reduced overall yields. Alternatively, oxidative cyclization strategies utilizing activated methyl-substituted heterocycles require stoichiometric amounts of oxidants and often necessitate elevated temperatures that can degrade sensitive functional groups. Furthermore, conventional carbonylation reactions typically depend on the use of toxic carbon monoxide gas supplied from high-pressure cylinders, posing significant safety risks and requiring specialized infrastructure that many contract manufacturing organizations lack. These limitations collectively result in increased production costs, extended lead times, and a restricted scope of compatible substrates, thereby hindering the rapid development of new drug candidates based on these privileged scaffolds.
The Novel Approach
In stark contrast to these legacy methods, the technology disclosed in CN115353511A employs a sophisticated transition metal-catalyzed tandem reaction that constructs the carbonyl-bridged framework in a single pot with remarkable efficiency. This novel approach utilizes a palladium catalyst system combined with a safe carbon monoxide surrogate generated in situ from a mixture of formic acid and acetic anhydride, effectively bypassing the need for external CO gas. The reaction proceeds at a mild temperature of 30°C, which preserves the integrity of sensitive functional groups and allows for the incorporation of diverse substituents on the aromatic rings. By merging trifluoroethylimidoyl chloride, propargylamine, and acrylamide derivatives, the process simultaneously forms multiple chemical bonds, including carbon-carbon and carbon-nitrogen linkages, through a cascade mechanism. This one-pot strategy not only simplifies the operational workflow by eliminating intermediate isolation steps but also significantly enhances the overall mass balance of the synthesis. The ability to tolerate a wide range of functional groups, including halogens and electron-withdrawing groups, makes this method particularly valuable for the late-stage functionalization of complex molecules.
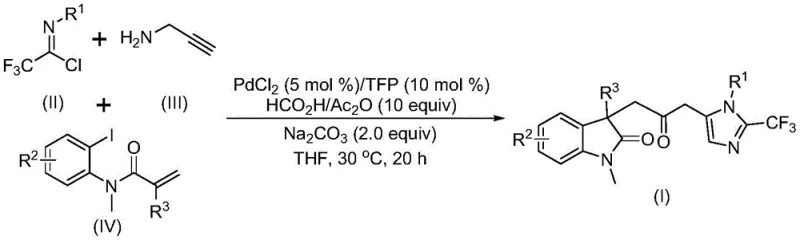
Mechanistic Insights into Pd-Catalyzed Carbonylation Cascade
The mechanistic pathway underpinning this transformation involves a intricate sequence of organometallic steps initiated by the oxidative addition of a zero-valent palladium species into the carbon-iodine bond of the acrylamide substrate. Following this activation, an intramolecular Heck-type reaction occurs to generate a divalent alkyl-palladium intermediate, which sets the stage for the subsequent carbonylation event. Crucially, the carbon monoxide required for this step is released slowly and steadily from the decomposition of the formic acid and acetic anhydride mixture, ensuring a controlled concentration of CO that prevents catalyst poisoning while driving the formation of the acyl-palladium species. Concurrently, a base-promoted intermolecular reaction between the trifluoroethylimidoyl chloride and propargylamine generates a trifluoroacetamidine intermediate, which undergoes isomerization to become reactive towards the palladium center. The final cyclization is triggered by the activation of this amidine species by the acyl-palladium intermediate, leading to the closure of the imidazole ring and the release of the final carbonyl-bridged biheterocyclic product. This elegant choreography of elementary steps highlights the dual role of the palladium catalyst in mediating both the carbonylation and the cyclization events, showcasing a high level of chemoselectivity.
From an impurity control perspective, the mild reaction conditions and the specific choice of ligands play a vital role in suppressing side reactions such as homocoupling or over-carbonylation. The use of tris(2-furyl)phosphine as a ligand stabilizes the palladium center and facilitates the reductive elimination step, which is often the rate-determining step in such cascades. Furthermore, the in situ generation of CO minimizes the risk of forming toxic metal-carbonyl clusters that can deactivate the catalyst or lead to heavy metal contamination in the final product. The compatibility of the reaction with various substituents on the aryl rings, as demonstrated by the successful synthesis of derivatives bearing chloro, fluoro, methyl, and trifluoromethyl groups, indicates a robust tolerance to electronic and steric variations. This broad substrate scope is essential for medicinal chemistry campaigns where rapid analog synthesis is required to optimize biological activity. The structural diversity achievable through this method is exemplified by the successful preparation of compounds I-1 through I-5, confirming the versatility of the protocol for generating libraries of biologically relevant scaffolds.
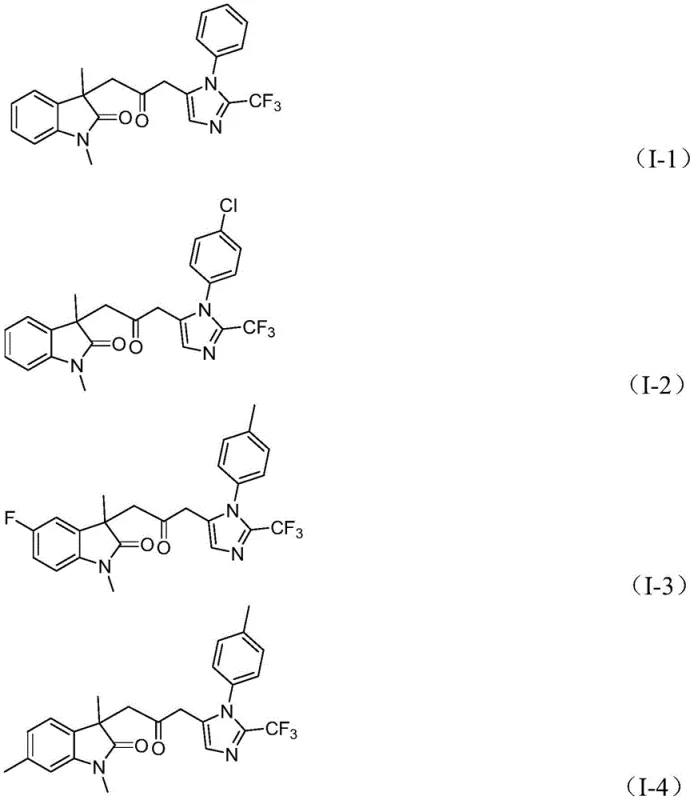
How to Synthesize Carbonyl-Bridged Biheterocyclic Compounds Efficiently
Implementing this synthesis route in a laboratory or pilot plant setting requires careful attention to reagent quality and reaction parameters to ensure optimal yields and reproducibility. The protocol is designed to be user-friendly, utilizing common organic solvents like tetrahydrofuran which effectively dissolve all reactants and facilitate homogeneous catalysis. The detailed standardized synthesis steps below outline the precise molar ratios and processing conditions necessary to achieve the high conversion rates reported in the patent data. Operators should ensure that the palladium catalyst and ligand are thoroughly mixed prior to the addition of substrates to maximize the formation of the active catalytic species. The reaction time window of 12 to 20 hours provides flexibility for monitoring reaction progress via TLC or HPLC, allowing for quenching once the starting material is fully consumed. Post-reaction workup involves standard filtration and silica gel treatment, followed by column chromatography, which are well-established techniques in process chemistry.
- Combine palladium chloride, tris(2-furyl)phosphine ligand, sodium carbonate, and the formic acid/acetic anhydride CO-source mixture in an organic solvent such as THF.
- Add the trifluoroethylimidoyl chloride, propargylamine, and acrylamide derivatives to the reaction vessel under inert atmosphere.
- Stir the mixture at 30°C for 12 to 20 hours, followed by filtration, silica gel treatment, and column chromatography purification.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain leaders, the adoption of this patented methodology offers substantial strategic benefits regarding cost structure and operational resilience. The elimination of high-pressure carbon monoxide gas removes the need for specialized storage facilities and safety monitoring systems, thereby drastically reducing the capital expenditure associated with setting up production lines for these intermediates. Moreover, the starting materials identified in the patent, such as propargylamine and various acrylamides, are commodity chemicals that are widely available from multiple global suppliers, mitigating the risk of supply chain disruptions caused by single-source dependencies. The mild reaction temperature of 30°C translates directly into lower energy consumption compared to traditional high-temperature cyclization processes, contributing to a reduced carbon footprint and lower utility costs per kilogram of product. Additionally, the high atom economy of the multicomponent reaction means that less raw material is wasted as byproducts, further enhancing the overall cost-efficiency of the manufacturing process. These factors collectively position this technology as a highly competitive option for the commercial production of complex pharmaceutical intermediates.
- Cost Reduction in Manufacturing: The substitution of toxic carbon monoxide gas with a liquid mixture of formic acid and acetic anhydride eliminates the logistical and safety costs associated with handling hazardous gases, leading to significant operational savings. By avoiding the need for high-pressure reactors, manufacturers can utilize standard glass-lined or stainless steel vessels, which lowers equipment maintenance and depreciation costs. The high yields reported in the experimental data suggest that raw material utilization is optimized, reducing the cost of goods sold for the final active pharmaceutical ingredient. Furthermore, the simplified one-pot nature of the reaction reduces labor hours and solvent usage associated with intermediate isolation and purification steps.
- Enhanced Supply Chain Reliability: The reliance on commercially available and inexpensive starting materials ensures a stable supply chain that is less vulnerable to geopolitical fluctuations or raw material shortages. The robustness of the reaction conditions allows for consistent batch-to-batch quality, which is critical for maintaining long-term contracts with pharmaceutical clients. The ability to scale the reaction to gram levels without loss of efficiency demonstrates that the process is ready for tech transfer to larger manufacturing sites, ensuring continuity of supply as demand grows. This reliability is further bolstered by the wide functional group tolerance, which allows for the sourcing of diverse substrate variants without needing to re-optimize the entire process for each new analog.
- Scalability and Environmental Compliance: The mild thermal profile of the reaction minimizes the generation of thermal waste and reduces the load on cooling systems, aligning with modern green chemistry principles. The use of standard organic solvents like THF, which can be recovered and recycled, supports sustainable manufacturing practices and helps facilities meet increasingly stringent environmental regulations. The absence of heavy metal contaminants in the final product, due to the efficient catalytic cycle, simplifies the downstream purification process and ensures compliance with strict residual metal limits imposed by regulatory agencies. This environmental compatibility makes the process attractive for companies aiming to improve their sustainability metrics while maintaining high production volumes.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis technology, derived directly from the patent specifications and experimental data. Understanding these details is crucial for R&D teams evaluating the feasibility of this route for their specific pipeline projects. The answers provided reflect the proven capabilities of the method as documented in the intellectual property, offering a clear picture of its operational boundaries and strengths. Clients are encouraged to review these points to assess how this technology aligns with their current manufacturing capabilities and quality standards.
Q: What are the safety advantages of this carbonylation method compared to traditional protocols?
A: This method utilizes a formic acid and acetic anhydride mixture as a carbon monoxide surrogate, eliminating the need for handling toxic, high-pressure CO gas cylinders, which significantly enhances operational safety in standard laboratory and pilot plant settings.
Q: Does this synthesis route support diverse substrate functionalization?
A: Yes, the protocol demonstrates excellent functional group tolerance, accommodating various substituents on the aryl rings including halogens, alkyl groups, alkoxy groups, and trifluoromethyl groups, allowing for the generation of a diverse library of biheterocyclic scaffolds.
Q: Is this process suitable for large-scale industrial production?
A: The patent explicitly confirms that the reaction conditions are mild (30°C) and the procedure has been successfully expanded to gram-scale reactions, indicating strong potential for commercial scale-up without requiring extreme pressure or temperature equipment.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Carbonyl-Bridged Biheterocyclic Compounds Supplier
As a premier CDMO partner, NINGBO INNO PHARMCHEM possesses the technical expertise and infrastructure required to translate this innovative patent technology into commercial reality for your organization. Our team has extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the transition from laboratory bench to industrial reactor is seamless and efficient. We maintain stringent purity specifications through our rigorous QC labs, guaranteeing that every batch of carbonyl-bridged biheterocyclic compounds meets the highest industry standards for pharmaceutical applications. Our commitment to quality and safety means that we can handle the specific reagents and conditions of this palladium-catalyzed process with the utmost professionalism and regulatory compliance. By partnering with us, you gain access to a supply chain that is both resilient and cost-effective, driven by our deep understanding of advanced organic synthesis.
We invite you to initiate a dialogue with our technical procurement team to discuss how we can tailor this synthesis route to your specific volume and purity requirements. Request a Customized Cost-Saving Analysis today to understand the potential economic impact of switching to this superior manufacturing method for your API intermediates. Our experts are ready to provide specific COA data and route feasibility assessments to support your decision-making process. Let us help you accelerate your drug development timeline with a reliable supply of high-quality heterocyclic building blocks that are synthesized using the latest advancements in catalytic chemistry.