Advanced Palladium-Catalyzed Synthesis of 2-Trifluoromethyl Imidazoles for Commercial Pharmaceutical Applications
Advanced Palladium-Catalyzed Synthesis of 2-Trifluoromethyl Imidazoles for Commercial Pharmaceutical Applications
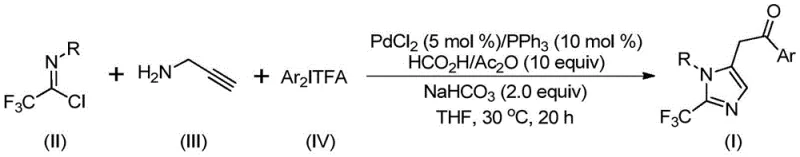
The strategic incorporation of trifluoromethyl groups into heterocyclic scaffolds represents a cornerstone of modern medicinal chemistry, profoundly enhancing the metabolic stability, lipophilicity, and bioavailability of drug candidates. Patent CN111423381B discloses a groundbreaking preparation method for 2-trifluoromethyl substituted imidazole compounds that addresses long-standing challenges in synthetic efficiency and safety. Unlike conventional approaches that often rely on hazardous or expensive trifluoromethylating agents, this novel protocol utilizes a palladium-catalyzed carbonylation cascade involving trifluoroethylimidoyl chloride, propargylamine, and diaryl iodonium salts. The reaction proceeds under remarkably mild conditions at 30°C, offering a robust pathway for generating high-value nitrogen-containing heterocycles. For R&D directors and procurement specialists seeking reliable pharmaceutical intermediate suppliers, this technology signifies a major leap forward in accessing complex fluorinated building blocks with superior purity profiles and reduced operational risks.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of trifluoromethyl-substituted nitrogen heterocycles has been plagued by significant technical and economic hurdles that impede efficient cost reduction in API manufacturing. Traditional methodologies frequently depend on unstable and potentially explosive reagents such as trifluorodiazoethane, which necessitates stringent safety protocols and specialized handling equipment, thereby inflating production costs. Furthermore, many existing routes suffer from poor atom economy, requiring harsh reaction conditions like extreme temperatures or strong bases that limit functional group tolerance. These limitations often result in complex purification processes to remove toxic byproducts and residual heavy metals, creating bottlenecks in the supply chain for high-purity OLED material or pharmaceutical precursors. The inability to easily diversify the substitution pattern on the imidazole ring without redesigning the entire synthetic sequence further restricts the utility of these older methods in rapid drug discovery campaigns.
The Novel Approach
The methodology described in the patent revolutionizes this landscape by employing a transition metal palladium-catalyzed intermolecular carbon-nitrogen bond formation strategy that is both operationally simple and highly versatile. By utilizing cheap and easily obtained trifluoroethyliminato chloride and propargylamine as starting materials, the process bypasses the need for dangerous diazo compounds entirely. The reaction leverages a unique carbonylation series where formic acid and acetic anhydride serve as safe carbon monoxide surrogates, eliminating the need for high-pressure CO gas cylinders. This approach not only enhances safety but also dramatically simplifies the reactor setup, making it ideal for commercial scale-up of complex polymer additives or fine chemicals. The broad substrate compatibility allows for the introduction of diverse aryl groups at the 1 and 5 positions of the imidazole ring, enabling medicinal chemists to rapidly explore structure-activity relationships without being constrained by synthetic feasibility.
Mechanistic Insights into Pd-Catalyzed Carbonylation Cascade
The catalytic cycle underpinning this transformation is a sophisticated orchestration of organometallic steps that ensures high regioselectivity and yield. Initially, an intermolecular carbon-nitrogen bond is formed between the trifluoroethylimidoyl chloride and propargylamine, promoted by the alkaline environment provided by sodium bicarbonate, to generate a trifluoroacetamidine intermediate. This species undergoes isomerization, setting the stage for the palladium catalyst to engage. The palladium center, coordinated by triphenylphosphine ligands, facilitates the palladation of the alkyne moiety, forming a crucial alkenyl palladium intermediate. Subsequent isomerization converts this into a more stable alkyl palladium species, which is primed for the carbonylation event. The carbon monoxide required for this step is generated in situ from the decomposition of formic acid in the presence of acetic anhydride, ensuring a steady, low-concentration supply of CO that drives the reaction forward without the risks associated with gaseous CO handling.
Following the insertion of carbon monoxide, an acyl palladium intermediate is formed, which then undergoes oxidative addition with the diaryl iodonium salt. This step generates a high-valent tetravalent palladium intermediate, a key feature that distinguishes this mechanism from standard cross-coupling reactions. The final step involves reductive elimination, which releases the desired 2-trifluoromethyl-substituted imidazole product and regenerates the active palladium(0) catalyst to continue the cycle. This intricate mechanism explains the exceptional functional group tolerance observed in the experimental data, as the mild conditions prevent the degradation of sensitive moieties like nitro or ester groups. For quality control teams, understanding this mechanism is vital for impurity control, as it highlights the importance of maintaining precise stoichiometry of the iodonium salt to prevent side reactions and ensure the stringent purity specifications required for clinical grade materials.
How to Synthesize 2-Trifluoromethyl Imidazole Efficiently
The practical execution of this synthesis is designed for maximum reproducibility and ease of handling, making it accessible for both laboratory scale optimization and pilot plant operations. The procedure involves charging a reaction vessel with a specific mixture of palladium chloride, triphenylphosphine, sodium bicarbonate, acetic anhydride, and formic acid in an aprotic organic solvent, preferably tetrahydrofuran (THF). To this catalytic mixture, the three key substrates—trifluoroethylimidoyl chloride, propargylamine, and diaryl iodonium salt—are added. The reaction is then allowed to proceed at a constant temperature of 30°C for a duration of 16 to 24 hours. This low-temperature requirement is a significant advantage, reducing energy consumption and thermal stress on the equipment. Upon completion, the workup is straightforward, involving simple filtration to remove inorganic salts followed by silica gel mixing and column chromatography purification.
- Charge a reaction vessel with palladium chloride, triphenylphosphine, sodium bicarbonate, acetic anhydride, and formic acid in an organic solvent such as THF.
- Add trifluoroethylimidoyl chloride, propargylamine, and diaryl iodonium salt to the mixture and stir at 30°C for 16 to 24 hours.
- Upon completion, filter the mixture, mix with silica gel, and purify via column chromatography to isolate the target 2-trifluoromethyl imidazole.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this patented methodology offers transformative benefits that directly impact the bottom line and operational resilience. The shift away from exotic, high-cost reagents to commodity chemicals like aniline derivatives and propargylamine drastically reduces the raw material cost base. Furthermore, the elimination of high-pressure gas handling and cryogenic conditions simplifies the infrastructure requirements for production facilities, allowing for faster deployment of manufacturing lines. The robustness of the reaction means fewer batch failures and less waste generation, contributing to a more sustainable and predictable supply chain. This reliability is critical for maintaining continuous production schedules for high-purity pharmaceutical intermediates, ensuring that downstream drug development programs are not delayed by material shortages.
- Cost Reduction in Manufacturing: The economic viability of this process is driven by the use of inexpensive catalysts and reagents that are commercially available in bulk quantities. By replacing hazardous trifluorodiazoethane with stable trifluoroethylimidoyl chloride, the process eliminates the need for specialized safety containment systems and expensive disposal protocols for toxic waste. The mild reaction temperature of 30°C significantly lowers energy consumption compared to traditional reflux conditions, resulting in substantial cost savings on utilities. Additionally, the high reaction efficiency and yields reported in the patent minimize the loss of valuable starting materials, optimizing the overall material balance and reducing the cost per kilogram of the final product.
- Enhanced Supply Chain Reliability: The reliance on widely available starting materials such as substituted anilines and phenylboronic acids (precursors to the iodonium salts) ensures a stable and diversified supply base. Unlike proprietary reagents that may be sourced from a single vendor, these commodities can be procured from multiple global suppliers, mitigating the risk of supply disruptions. The simplicity of the post-treatment process, which avoids complex extractions or distillations, shortens the production cycle time, allowing for faster turnaround on orders. This agility enables manufacturers to respond more quickly to fluctuating market demands and urgent requests from R&D departments for new compound libraries.
- Scalability and Environmental Compliance: The protocol has been demonstrated to be expandable to the gram level with consistent results, indicating a clear path towards multi-kilogram and ton-scale production. The use of THF as a solvent, while requiring recovery systems, is well-established in the industry, and the absence of heavy metal contaminants in the final product simplifies the purification burden. The generation of benign byproducts and the avoidance of stoichiometric oxidants or reductants align with green chemistry principles, facilitating easier regulatory approval and environmental compliance. This scalability ensures that the method can support the entire lifecycle of a drug candidate, from early-stage discovery to commercial launch, without the need for process redevelopment.
Frequently Asked Questions (FAQ)
The following questions address common technical inquiries regarding the implementation and scope of this synthesis technology. These insights are derived directly from the experimental data and mechanistic understanding provided in the patent documentation. Understanding these details is essential for process chemists evaluating the feasibility of integrating this route into their existing manufacturing workflows.
Q: What are the primary advantages of this palladium-catalyzed method over traditional trifluoromethylation routes?
A: This method utilizes cheap and readily available starting materials like trifluoroethylimidoyl chloride instead of hazardous trifluorodiazoethane. It operates under extremely mild conditions (30°C) and demonstrates excellent functional group tolerance, allowing for the synthesis of diverse derivatives without harsh reagents.
Q: What is the substrate scope for the aryl groups in this synthesis?
A: The reaction exhibits broad substrate compatibility. Both the imidoyl chloride component (R group) and the diaryl iodonium salt (Ar group) can bear various substituents including methyl, tert-butyl, halogens (chloro, bromo, fluoro), trifluoromethyl, nitro, and methoxy groups at ortho, meta, or para positions, yielding products with high efficiency.
Q: Is this process suitable for large-scale industrial production?
A: Yes, the patent explicitly states the method is expandable to the gram level and provides possibilities for industrial large-scale production. The use of inexpensive catalysts (PdCl2), simple post-treatment (filtration and chromatography), and mild reaction temperatures significantly lowers the barrier for commercial scale-up compared to cryogenic or high-pressure alternatives.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 2-Trifluoromethyl Imidazole Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical role that advanced fluorination technologies play in accelerating drug discovery and development. Our team of expert process chemists possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your project transitions smoothly from the bench to the plant. We are committed to delivering high-purity intermediates that meet stringent purity specifications, supported by our rigorous QC labs equipped with state-of-the-art analytical instrumentation. Whether you require custom synthesis of novel imidazole derivatives or large-scale supply of established building blocks, our infrastructure is designed to support your most demanding requirements with speed and precision.
We invite you to collaborate with us to leverage this cutting-edge palladium-catalyzed technology for your next project. Contact our technical procurement team today to request a Customized Cost-Saving Analysis tailored to your specific volume needs. We are ready to provide specific COA data and comprehensive route feasibility assessments to demonstrate how our manufacturing capabilities can enhance your supply chain efficiency and reduce your overall development costs.