Scalable Synthesis of Fasudil Hydrochloride: Overcoming Impurity Challenges in Rho-Kinase Inhibitor Manufacturing
The pharmaceutical landscape for cardiovascular therapeutics continues to demand higher standards of purity and process efficiency, particularly for critical vasodilators like Fasudil Hydrochloride. Patent CN102120739A introduces a transformative preparation method that addresses long-standing synthetic bottlenecks associated with this Rho-kinase inhibitor. Traditionally, the synthesis of Fasudil Hydrochloride, chemically known as hexahydro-1-(5-isoquinolinesulfonyl)-1H-1,4-diazepine hydrochloride, has been plagued by selectivity issues that compromise yield and complicate downstream purification. This novel technical disclosure outlines a robust route utilizing N-protected homopiperazine as a key strategic intermediate. By masking one of the reactive nitrogen centers prior to sulfonylation, the process effectively prevents the formation of difficult-to-remove bis-sulfonylated byproducts. For R&D directors and process chemists, this represents a significant leap forward in impurity control, offering a pathway to achieve exceptional purity profiles, such as the 99.99% demonstrated in the patent examples, which is essential for meeting stringent regulatory requirements for injectable or oral cardiovascular medications.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
In the established prior art, the synthesis of Fasudil Hydrochloride typically involves the direct reaction of 5-isoquinolinesulfonyl chloride with unprotected homopiperazine. While conceptually straightforward, this approach suffers from inherent chemoselectivity defects that severely impact commercial viability. Homopiperazine possesses two secondary amine groups with similar nucleophilic reactivity. Consequently, when exposed to the electrophilic sulfonyl chloride, there is a high probability of double substitution, leading to the formation of a bis-sulfonylated impurity, often referred to as Compound 10 in technical literature. This side reaction not only consumes valuable starting materials but also generates a byproduct that is structurally similar to the target molecule, making separation via standard crystallization or chromatography exceptionally difficult and costly. Furthermore, the commercial availability of high-purity unprotected homopiperazine is often limited, and its market price is prohibitively high, directly inflating the cost of goods sold (COGS) for the final API. The accumulation of these inefficiencies results in lower overall yields and a final product that may require extensive, yield-eroding purification steps to meet pharmacopeial standards.
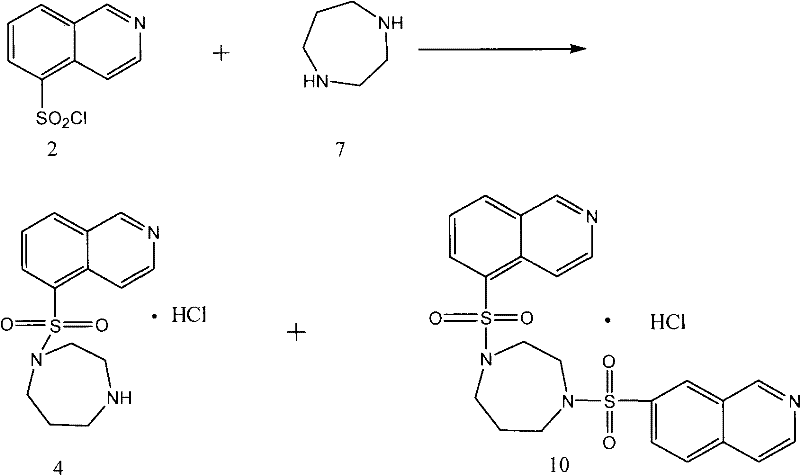
The Novel Approach
The innovative methodology disclosed in the patent circumvents these challenges by employing a protective group strategy that fundamentally alters the reaction trajectory. Instead of using the naked diamine, the process utilizes an N-protected homopiperazine derivative where one nitrogen atom is temporarily masked with a stable protecting group, such as a benzyloxycarbonyl (Cbz) or tert-butoxycarbonyl (Boc) moiety. This modification ensures that only the single free amine group is available for nucleophilic attack on the 5-isoquinolinesulfonyl chloride. As a result, the sulfonylation proceeds with high regioselectivity, virtually eliminating the formation of the bis-sulfonylated impurity. Following the coupling reaction, the protecting group is cleanly removed under acidic hydrolysis conditions to reveal the final active pharmaceutical ingredient. This two-stage sequence—protected coupling followed by deprotection—not only simplifies the reaction mixture but also facilitates easier purification, as the intermediate and final product have distinct physicochemical properties compared to the potential byproducts of the unprotected route.
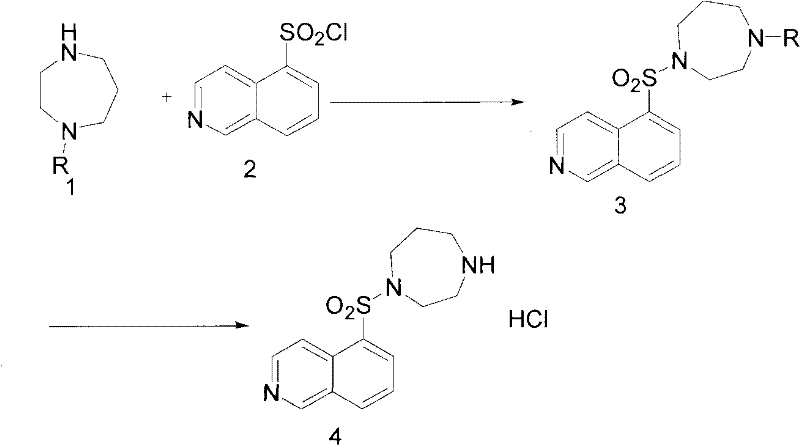
Mechanistic Insights into Protective Group Strategy and Rearrangement
A critical component of this cost-effective synthesis is the in-house generation of the N-protected homopiperazine starting material from inexpensive commodity chemicals. The patent details a sophisticated four-step sequence beginning with 4-piperidone hydrochloride hydrate, a widely available and low-cost feedstock. The mechanism initiates with the protection of the piperidine nitrogen, followed by an oximation reaction to form the corresponding ketoxime. The pivotal step in this sequence is the Beckmann rearrangement, a molecular transposition triggered by reagents like tosyl chloride under basic conditions. This rearrangement expands the six-membered piperidine ring into the seven-membered diazepine skeleton required for the Fasudil structure, simultaneously installing the second nitrogen atom necessary for the homopiperazine core. This mechanistic elegance allows manufacturers to bypass the supply chain risks associated with purchasing complex heterocycles directly. The final reduction of the carbonyl group yields the fully protected homopiperazine, ready for the sulfonylation step. By controlling the stereochemistry and reaction conditions during the rearrangement and reduction phases, manufacturers can minimize the formation of geometric isomers or over-reduced species, ensuring a clean input for the final coupling reaction.
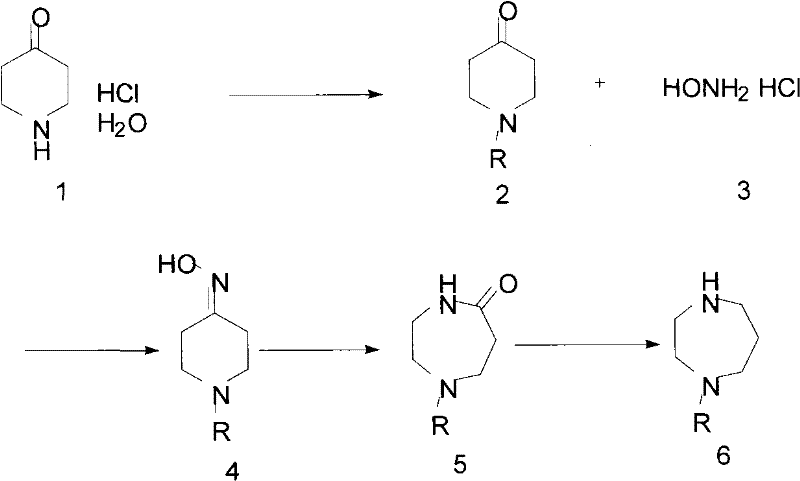
From an impurity control perspective, the choice of the protecting group is paramount to the success of the overall process. The patent highlights several viable options, including benzoyl, benzyloxycarbonyl, and tert-butoxycarbonyl groups, each offering distinct advantages regarding stability and removal conditions. For instance, the benzyloxycarbonyl group provides excellent stability during the sulfonylation step but can be efficiently cleaved using concentrated hydrochloric acid under reflux conditions. This deprotection step is designed to be orthogonal to the sulfonamide bond, ensuring that the core structure of the Fasudil molecule remains intact while the protecting group is quantitatively removed. The resulting crude product, after neutralization and extraction, is subjected to recrystallization from solvents such as ethanol or mixtures of ethanol and water. This final purification step leverages the high crystallinity of the Fasudil hydrochloride salt to exclude any trace organic impurities or residual starting materials. The combination of selective synthesis and robust crystallization creates a powerful barrier against impurity ingress, resulting in a product profile that consistently meets the rigorous demands of global regulatory agencies for cardiovascular therapeutics.
How to Synthesize Fasudil Hydrochloride Efficiently
The execution of this synthesis requires precise control over reaction parameters to maximize the benefits of the protective group strategy. The process begins with the preparation of the N-protected homopiperazine, which serves as the foundational building block for the entire sequence. Once this intermediate is secured, the sulfonylation reaction is conducted in a biphasic or homogeneous organic system, typically using dichloromethane as the solvent and an organic base like triethylamine or pyridine to scavenge the generated hydrogen chloride. Maintaining the temperature between 15°C and 30°C during this exothermic coupling is crucial to prevent thermal degradation or side reactions. Following the coupling, the workup involves aqueous extraction to remove inorganic salts, followed by the critical deprotection step using strong acid. The detailed standardized operating procedures for each unit operation, including specific molar ratios, solvent volumes, and agitation speeds, are essential for reproducibility at scale.
- Synthesize N-protected homopiperazine starting from 4-piperidone hydrochloride hydrate via protection, oximation, Beckmann rearrangement, and reduction.
- Perform sulfonylation by reacting 5-isoquinolinesulfonyl chloride with the N-protected homopiperazine in an organic solvent like dichloromethane.
- Remove the amino protective group via acid hydrolysis (e.g., concentrated HCl) and purify the final Fasudil Hydrochloride through recrystallization.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain leaders, the adoption of this patented methodology offers substantial strategic benefits beyond mere technical superiority. The primary advantage lies in the decoupling of the supply chain from expensive, specialized raw materials. By synthesizing the homopiperazine core from 4-piperidone, a bulk commodity chemical, manufacturers can significantly reduce their exposure to price volatility in the fine chemical market. This vertical integration of the synthesis route ensures a more predictable cost structure and reduces dependency on single-source suppliers for complex heterocycles. Furthermore, the elimination of the bis-sulfonylated byproduct drastically simplifies the purification train. In conventional processes, removing structurally similar impurities often requires multiple recrystallizations or preparative chromatography, both of which are time-consuming and result in significant yield loss. By preventing the formation of these impurities at the source, the new process enhances the overall mass balance, leading to higher throughput and reduced waste generation, which translates directly into lower manufacturing costs per kilogram of active ingredient.
- Cost Reduction in Manufacturing: The economic impact of this process is driven by the substitution of high-cost unprotected homopiperazine with low-cost 4-piperidone derivatives. Additionally, the high selectivity of the protected route minimizes the loss of the valuable isoquinoline sulfonyl chloride fragment, which is often the most expensive reagent in the synthesis. By avoiding the formation of the bis-sulfonylated waste product, the effective utilization of raw materials is maximized. This efficiency gain means that less raw material is required to produce the same amount of final API, thereby lowering the variable cost of production. Moreover, the simplified purification protocol reduces the consumption of solvents and energy associated with extensive recrystallization cycles, contributing to a leaner and more cost-effective manufacturing operation that improves margin potential in a competitive generic drug market.
- Enhanced Supply Chain Reliability: Relying on commodity feedstocks like 4-piperidone hydrochloride hydrate significantly de-risks the supply chain compared to sourcing specialized diamines. Commodity chemicals are produced by multiple vendors globally, ensuring continuity of supply even if one supplier faces disruptions. This diversification of the raw material base provides procurement teams with greater negotiating leverage and flexibility. Additionally, the robustness of the synthetic route, characterized by standard unit operations such as sulfonylation, hydrolysis, and crystallization, makes it highly adaptable to different manufacturing sites. This flexibility allows for multi-site production strategies, further securing the supply of this critical cardiovascular intermediate against regional logistical challenges or geopolitical instabilities that might affect the distribution of more exotic reagents.
- Scalability and Environmental Compliance: The process is inherently designed for scalability, utilizing reaction conditions that are easily transferable from pilot plant to commercial-scale reactors. The use of common solvents like dichloromethane, ethyl acetate, and ethanol aligns with standard solvent recovery infrastructure found in most GMP facilities. From an environmental perspective, the high atom economy resulting from the selective reaction reduces the volume of hazardous waste generated per unit of product. The avoidance of difficult-to-separate byproducts means less mother liquor waste containing complex organic impurities, simplifying wastewater treatment protocols. This alignment with green chemistry principles not only reduces disposal costs but also supports corporate sustainability goals, making the manufacturing process more attractive to environmentally conscious stakeholders and regulatory bodies focused on reducing the pharmaceutical industry's ecological footprint.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this advanced synthesis route. These insights are derived directly from the experimental data and process descriptions within the patent documentation, providing a clear understanding of the operational realities and benefits. Understanding these nuances is vital for technical teams evaluating the feasibility of technology transfer and for commercial teams assessing the value proposition of this manufacturing method in the context of global supply chains.
Q: Why is N-protected homopiperazine used instead of direct homopiperazine?
A: Direct reaction of homopiperazine with 5-isoquinolinesulfonyl chloride often leads to bis-sulfonylation byproducts (Compound 10) because both amine groups are reactive. Using an N-protected variant ensures mono-sulfonylation, significantly improving yield and simplifying purification.
Q: What purity levels can be achieved with this method?
A: The patented method utilizing N-protected intermediates and subsequent recrystallization allows for the achievement of extremely high purity levels, specifically cited as 99.99% in experimental examples, which is critical for pharmaceutical grade API intermediates.
Q: Is the starting material 4-piperidone cost-effective?
A: Yes, 4-piperidone hydrochloride hydrate is a readily available commodity chemical. Synthesizing the homopiperazine core from this precursor is generally more cost-competitive than purchasing expensive, unprotected homopiperazine directly, especially when factoring in the yield losses of conventional methods.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Fasudil Hydrochloride Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of high-purity intermediates in the development of life-saving cardiovascular therapies. Our technical team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the intricate protective group chemistry described in this patent is executed with precision and consistency. We understand that achieving the stringent purity specifications required for Fasudil Hydrochloride demands rigorous QC labs and state-of-the-art analytical capabilities. Our facility is equipped to handle the multi-step synthesis of N-protected homopiperazine and the subsequent sulfonylation with the highest standards of quality control, guaranteeing a product that meets or exceeds the 99.99% purity benchmarks set forth in the latest technical literature.
We invite pharmaceutical partners to collaborate with us to leverage this superior manufacturing technology for your supply chain. By choosing NINGBO INNO PHARMCHEM, you gain access to a Customized Cost-Saving Analysis that quantifies the economic benefits of switching to this protected intermediate route versus traditional methods. We encourage you to contact our technical procurement team today to request specific COA data from our recent batches and to discuss route feasibility assessments tailored to your specific volume requirements. Let us help you secure a reliable, cost-effective, and high-quality supply of Fasudil Hydrochloride for your global markets.