Advanced Palladium-Catalyzed Synthesis of 2-Trifluoromethyl Imidazoles for Commercial Pharmaceutical Applications
Advanced Palladium-Catalyzed Synthesis of 2-Trifluoromethyl Imidazoles for Commercial Pharmaceutical Applications
The integration of trifluoromethyl groups into nitrogen-containing heterocycles represents a cornerstone strategy in modern medicinal chemistry, significantly enhancing the metabolic stability, lipophilicity, and bioavailability of drug candidates. As highlighted in patent CN111423381A, the development of efficient synthetic routes for 2-trifluoromethyl substituted imidazole compounds addresses a critical bottleneck in the supply chain for high-value pharmaceutical intermediates. These scaffolds are not merely academic curiosities but are foundational structures found in a vast array of bioactive molecules, ranging from potent kinase inhibitors to antifungal agents, as illustrated by the structural diversity seen in known bioactive compounds. 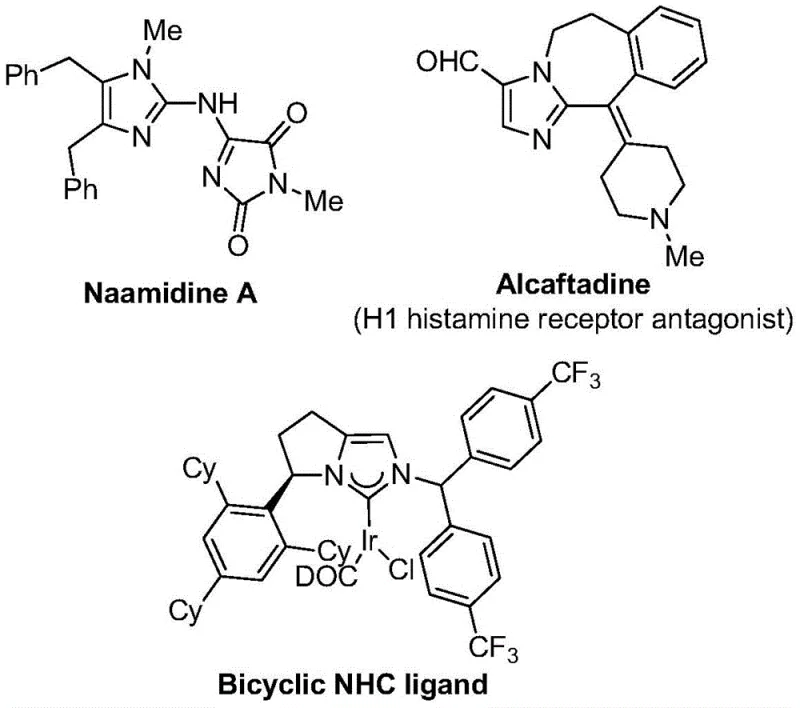 . The ability to access these motifs through a robust, scalable, and cost-effective methodology is paramount for R&D directors seeking to accelerate lead optimization and for procurement teams aiming to secure reliable sources of complex building blocks.
. The ability to access these motifs through a robust, scalable, and cost-effective methodology is paramount for R&D directors seeking to accelerate lead optimization and for procurement teams aiming to secure reliable sources of complex building blocks.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the construction of trifluoromethylated imidazole rings has relied heavily on the use of specialized trifluoromethyl synthons such as trifluorodiazoethane, which pose significant safety hazards due to their explosive nature and instability. Alternative approaches often require harsh reaction conditions, including elevated temperatures and strong bases, which can lead to poor functional group tolerance and the formation of complex impurity profiles that are difficult to remove. Furthermore, many traditional methods suffer from limited substrate scope, failing to accommodate electron-deficient or sterically hindered aryl groups, thereby restricting the chemical space available for drug design. These limitations translate directly into increased manufacturing costs and extended lead times, as extensive purification steps and specialized safety protocols become necessary to handle reactive intermediates safely.
The Novel Approach
In stark contrast, the methodology disclosed in CN111423381A introduces a transformative transition metal palladium-catalyzed carbonylation cascade reaction that operates under remarkably mild conditions. By utilizing cheap and readily available starting materials—specifically trifluoroethylimidoyl chloride, propargylamine, and diaryliodonium salts—this novel route bypasses the need for dangerous diazo compounds entirely. The reaction proceeds efficiently at a low temperature of 30°C, utilizing a formic acid and acetic anhydride mixture as a safe carbon monoxide surrogate, which eliminates the need for handling toxic CO gas cylinders. 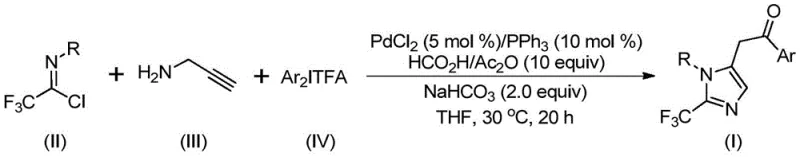 . This approach not only simplifies the operational complexity but also dramatically broadens the utility of the method, allowing for the synthesis of diversely substituted imidazole derivatives that were previously inaccessible or economically unviable to produce.
. This approach not only simplifies the operational complexity but also dramatically broadens the utility of the method, allowing for the synthesis of diversely substituted imidazole derivatives that were previously inaccessible or economically unviable to produce.
Mechanistic Insights into Palladium-Catalyzed Carbonylation Cascade
The elegance of this synthesis lies in its intricate catalytic cycle, which orchestrates multiple bond-forming events in a single pot. The mechanism is proposed to initiate with a base-promoted intermolecular carbon-nitrogen bond formation between the trifluoroethylimidoyl chloride and propargylamine, generating a trifluoroacetamidine intermediate. This species subsequently undergoes isomerization and palladium-catalyzed aminopalladation of the alkyne moiety to form a vinyl-palladium intermediate. A crucial step follows where isomerization yields an alkyl-palladium species, which then engages in a carbonylation reaction driven by carbon monoxide released in situ from the formic acid/acetic anhydride system. This generates an acyl-palladium intermediate that is poised for the final C-C bond formation.
The cycle culminates in an oxidative addition of the diaryliodonium salt to the palladium center, forming a high-valent tetravalent palladium intermediate, a rare and powerful mechanistic feature that enables the transfer of the aryl group. Finally, reductive elimination releases the desired 2-trifluoromethyl substituted imidazole product and regenerates the active palladium catalyst. This sophisticated mechanism ensures high atom economy and exceptional selectivity, minimizing the formation of side products. The mild reaction temperature of 30°C further suppresses thermal degradation pathways, ensuring that the final crude product possesses a high degree of purity, which significantly reduces the burden on downstream purification processes and aligns with the stringent quality requirements of pharmaceutical manufacturing.
How to Synthesize 2-Trifluoromethyl Imidazole Efficiently
The practical implementation of this synthesis is designed for ease of execution in both laboratory and pilot plant settings. The protocol involves a straightforward one-pot procedure where all reagents, including the palladium catalyst, ligand, base, and substrates, are combined in an aprotic organic solvent such as tetrahydrofuran (THF). The reaction mixture is stirred at 30°C for a period of 16 to 24 hours, allowing sufficient time for the cascade transformation to reach completion. Upon conclusion of the reaction, the workup is uncomplicated, involving simple filtration to remove inorganic salts followed by silica gel mixing and column chromatography purification.
- Combine palladium chloride, triphenylphosphine, sodium bicarbonate, and a formic acid/acetic anhydride mixture in an organic solvent such as THF.
- Add trifluoroethylimidoyl chloride, propargylamine, and diaryliodonium salt to the reaction mixture under stirring.
- Maintain the reaction at 30°C for 16 to 24 hours, then filter and purify via column chromatography to isolate the target compound.
Commercial Advantages for Procurement and Supply Chain Teams
From a commercial perspective, this patented technology offers compelling advantages that directly address the pain points of cost, reliability, and scalability in the supply of fine chemical intermediates. The shift towards using commodity chemicals as starting materials fundamentally alters the cost structure of producing these valuable heterocycles, making them accessible for larger volume applications without compromising on quality or purity standards required for GMP environments.
- Cost Reduction in Manufacturing: The economic viability of this process is underpinned by the use of inexpensive and commercially abundant reagents. Trifluoroethylimidoyl chloride and propargylamine are commodity chemicals available from multiple global suppliers, mitigating the risk of single-source dependency and price volatility. Furthermore, the catalytic system employs palladium chloride and triphenylphosphine, which are significantly more cost-effective than specialized ligands or exotic metal complexes often required in cross-coupling reactions. The elimination of hazardous reagents like trifluorodiazoethane also removes the need for expensive safety infrastructure and waste disposal protocols associated with explosive materials, leading to substantial indirect cost savings in facility operations and regulatory compliance.
- Enhanced Supply Chain Reliability: The robustness of the reaction conditions contributes directly to supply chain stability. Operating at a mild 30°C reduces the energy consumption compared to high-temperature reflux processes, lowering the operational expenditure for manufacturing partners. The high substrate compatibility means that a single standardized protocol can be adapted to produce a wide library of analogues by simply varying the aryl groups on the starting materials. This flexibility allows manufacturers to respond rapidly to changing demand for specific derivatives without needing to revalidate entirely new processes, thereby reducing lead times for high-purity pharmaceutical intermediates and ensuring continuity of supply for downstream API production.
- Scalability and Environmental Compliance: The process has been demonstrated to be scalable, with the patent explicitly noting its potential for expansion to gram-level and beyond, paving the way for multi-kilogram commercial production. The use of a carbon monoxide surrogate (formic acid/acetic anhydride) avoids the logistical and safety challenges of storing and transporting high-pressure CO gas, simplifying the engineering requirements for scale-up. Additionally, the reaction exhibits high atom efficiency and generates minimal hazardous waste, aligning with green chemistry principles. This environmental profile facilitates easier permitting and regulatory approval for manufacturing sites, ensuring long-term sustainability and compliance with increasingly strict environmental regulations in the chemical industry.
Frequently Asked Questions (FAQ)
To assist technical decision-makers in evaluating this technology for their specific applications, we have compiled answers to common inquiries regarding the operational parameters and scope of this synthesis method. These insights are derived directly from the experimental data and technical specifications outlined in the patent documentation, providing a clear picture of what can be expected during technology transfer.
Q: What are the optimal reaction conditions for this synthesis?
A: The patent specifies a mild temperature of 30°C with a reaction time of 16 to 24 hours in THF solvent, utilizing PdCl2 and PPh3 as the catalytic system.
Q: Does this method tolerate diverse functional groups?
A: Yes, the methodology demonstrates excellent substrate compatibility, successfully accommodating substituents such as methyl, tert-butyl, halogens, trifluoromethyl, and nitro groups on the aryl rings.
Q: Is this process suitable for large-scale manufacturing?
A: The process is designed for scalability, utilizing cheap and readily available starting materials and operating under mild conditions that facilitate safe expansion to industrial production levels.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 2-Trifluoromethyl Imidazole Supplier
At NINGBO INNO PHARMCHEM, we recognize the strategic importance of accessing advanced synthetic methodologies to maintain a competitive edge in the pharmaceutical market. Our team of expert chemists has thoroughly analyzed the potential of the palladium-catalyzed carbonylation route described in CN111423381A and is fully prepared to implement this technology for your projects. We possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your transition from benchtop discovery to commercial supply is seamless. Our state-of-the-art facilities are equipped with rigorous QC labs capable of meeting stringent purity specifications, guaranteeing that every batch of 2-trifluoromethyl imidazole intermediate meets the highest industry standards.
We invite you to leverage our technical expertise to optimize your supply chain and reduce your overall cost of goods. By partnering with us, you gain access to a Customized Cost-Saving Analysis tailored to your specific volume requirements and purity needs. We encourage you to contact our technical procurement team today to request specific COA data for related intermediates and to discuss route feasibility assessments for your target molecules. Let us help you secure a reliable supply of these critical building blocks and accelerate your drug development timeline.