Scalable Metal-Free Synthesis of Pyrimidoindazole Compounds for Commercial API Production
Introduction to Advanced Pyrimidoindazole Synthesis
The development of efficient synthetic routes for bioactive heterocycles remains a cornerstone of modern pharmaceutical research. Patent CN109912606B introduces a groundbreaking methodology for the synthesis of pyrimidoindazole compounds, a privileged scaffold known for its potent biological activity as protein kinase inhibitors and therapeutic agents for neurodegenerative diseases. This technology represents a significant leap forward in organic synthesis, transitioning away from resource-intensive multi-step protocols toward a streamlined, metal-free one-pot tandem reaction. By leveraging inexpensive reagents such as ammonium iodide and triethylamine, this process addresses critical pain points in API intermediate manufacturing, including high catalyst costs and complex waste streams. The ability to directly construct the pyrimidoindazole core from simple aromatic aldehydes and 3-aminoindazoles under oxidative conditions offers a robust platform for generating diverse chemical libraries.
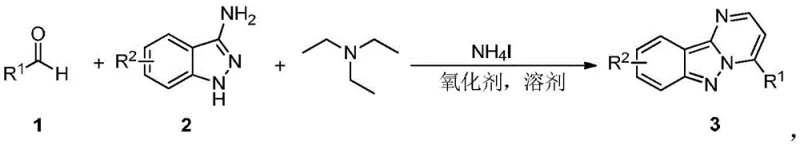
Furthermore, the operational simplicity of this protocol cannot be overstated. Traditional methods often require stringent anhydrous conditions, inert atmospheres, and specialized equipment to handle sensitive organometallic catalysts. In contrast, this novel approach tolerates open-air conditions when using molecular oxygen or air as the terminal oxidant, drastically simplifying the engineering requirements for scale-up. The reaction proceeds efficiently at temperatures ranging from 110°C to 150°C, providing a balance between reaction kinetics and energy consumption that is ideal for commercial production. For R&D teams seeking to optimize lead compounds, this method provides rapid access to structural analogs, accelerating the drug discovery timeline while maintaining high standards of purity and yield.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the construction of the pyrimidoindazole skeleton has relied on condensation reactions involving 3-aminoindazoles and 1,3-dicarbonyl compounds or propiolates, often necessitating harsh acidic or basic conditions. Alternative strategies such as Grieco condensation or Michael addition-cyclization sequences frequently employ corrosive reagents and expensive transition metal catalysts like palladium or copper complexes. These conventional pathways suffer from significant drawbacks, including low atom economy, the generation of hazardous heavy metal waste, and the need for rigorous purification steps to remove trace metal residues to meet pharmaceutical regulatory standards. Moreover, the multi-step nature of these traditional syntheses leads to cumulative yield losses and increased production timelines, creating bottlenecks in the supply chain for high-purity pharmaceutical intermediates.
The Novel Approach
The methodology disclosed in CN109912606B fundamentally reimagines this synthetic challenge by utilizing a transition-metal-free catalytic system driven by ammonium iodide and triethylamine. This innovative route bypasses the need for pre-functionalized substrates or precious metal catalysts, instead relying on an oxidative cyclization mechanism that directly couples aromatic aldehydes with 3-aminoindazoles. The use of triethylamine serves a dual purpose as both a base and a reactant, significantly lowering the raw material costs compared to specialized ligands or organometallic reagents. Additionally, the flexibility in oxidant selection—ranging from organic peroxides like DTBP and BPO to green oxidants like air and oxygen—allows manufacturers to tailor the process for either maximum yield or minimum environmental footprint. This adaptability makes the novel approach superior in terms of both economic viability and sustainability.
Mechanistic Insights into NH4I-Catalyzed Oxidative Cyclization
The core of this transformation lies in the synergistic interaction between ammonium iodide and the oxidant to generate reactive iodine species in situ, which facilitate the activation of the aldehyde and subsequent cyclization. Mechanistically, the reaction likely proceeds through an initial condensation between the amino group of the indazole and the carbonyl of the aldehyde, followed by iodine-mediated oxidation to form the requisite C-N and C-C bonds that close the pyrimidine ring. The absence of transition metals eliminates the risk of metal-induced side reactions or catalyst deactivation by substrate coordinating groups, ensuring consistent performance across a wide range of electronic environments. This mechanistic clarity allows chemists to predict reaction outcomes with greater confidence, reducing the empirical trial-and-error often associated with developing new synthetic routes for complex heterocycles.
From an impurity control perspective, the mild reaction conditions and the specific selectivity of the iodine-mediated oxidation contribute to a cleaner crude reaction profile. Unlike strong acid or base catalyzed condensations that can lead to polymerization or decomposition of sensitive functional groups, this protocol preserves delicate moieties such as halogens and nitro groups. The use of triethylamine helps to neutralize acidic byproducts generated during the oxidation cycle, further stabilizing the reaction mixture. Consequently, the downstream purification process is simplified, often requiring only standard silica gel chromatography or recrystallization to achieve high-purity products suitable for biological testing. This level of control over the impurity profile is critical for meeting the stringent quality specifications required for clinical grade intermediates.
How to Synthesize Pyrimidoindazole Efficiently
To implement this synthesis effectively, operators should dissolve the aromatic aldehyde, 3-aminoindazole, and triethylamine in a high-boiling solvent such as chlorobenzene or toluene. The addition of ammonium iodide and the chosen oxidant initiates the cascade, which is typically maintained at 120°C for 12 hours to ensure complete conversion. Detailed standardized synthesis steps see the guide below.
- Dissolve aromatic aldehyde, 3-aminoindazole, and triethylamine in a suitable solvent such as chlorobenzene or toluene.
- Add ammonium iodide (NH4I) and a selected oxidant (e.g., air, oxygen, or organic peroxides) to the reaction mixture.
- Heat the mixture to 110-150°C and stir for approximately 12 hours to complete the cyclization, followed by standard aqueous workup and purification.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this metal-free synthesis offers tangible benefits that extend beyond the laboratory bench. The primary advantage lies in the drastic simplification of the raw material portfolio. By replacing expensive transition metal catalysts and specialized ligands with commodity chemicals like ammonium iodide and triethylamine, the direct material costs are significantly reduced. Furthermore, the elimination of heavy metals removes the need for costly scavenging resins and extensive analytical testing for residual metals, streamlining the quality control workflow. This reduction in process complexity translates directly into lower operating expenses and a more predictable cost structure for long-term manufacturing contracts.
- Cost Reduction in Manufacturing: The substitution of precious metal catalysts with inexpensive ammonium salts and amines results in substantial cost savings per kilogram of product. Additionally, the one-pot nature of the reaction minimizes solvent usage and energy consumption associated with intermediate isolation and purification steps. The ability to use air or oxygen as a stoichiometric oxidant further reduces reagent costs compared to proprietary organic oxidants, making the overall process economically attractive for large-scale production of pharmaceutical intermediates.
- Enhanced Supply Chain Reliability: The reliance on widely available bulk chemicals ensures a stable and resilient supply chain. Unlike specialized catalysts that may face sourcing bottlenecks or long lead times, reagents like triethylamine and aromatic aldehydes are produced globally in massive quantities. This availability mitigates the risk of production delays due to raw material shortages. Moreover, the robustness of the reaction conditions allows for flexible manufacturing scheduling, as the process is less sensitive to minor variations in temperature or atmosphere compared to sensitive organometallic couplings.
- Scalability and Environmental Compliance: The process is inherently scalable, having been demonstrated in various solvent systems and oxidant conditions that can be adapted to different reactor configurations. The avoidance of toxic heavy metals aligns with increasingly strict environmental regulations regarding waste disposal and effluent treatment. By generating less hazardous waste and utilizing greener oxidants like air, manufacturers can reduce their environmental compliance burden and enhance their corporate sustainability profiles, which is a growing priority for global pharmaceutical partners.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding this synthesis technology. These answers are derived directly from the experimental data and technical specifications outlined in the patent documentation, providing a reliable basis for feasibility assessments.
Q: What are the key advantages of this metal-free synthesis method over traditional routes?
A: This method eliminates the need for expensive and toxic transition metal catalysts, utilizes inexpensive triethylamine, and operates under mild conditions with a wide substrate scope, significantly reducing environmental impact and production costs.
Q: Can this process be scaled for industrial manufacturing of kinase inhibitor intermediates?
A: Yes, the process is designed for industrial production. It uses simple raw materials, avoids complex multi-step purifications, and allows for open-air or sealed conditions depending on the oxidant, making it highly scalable and operationally convenient.
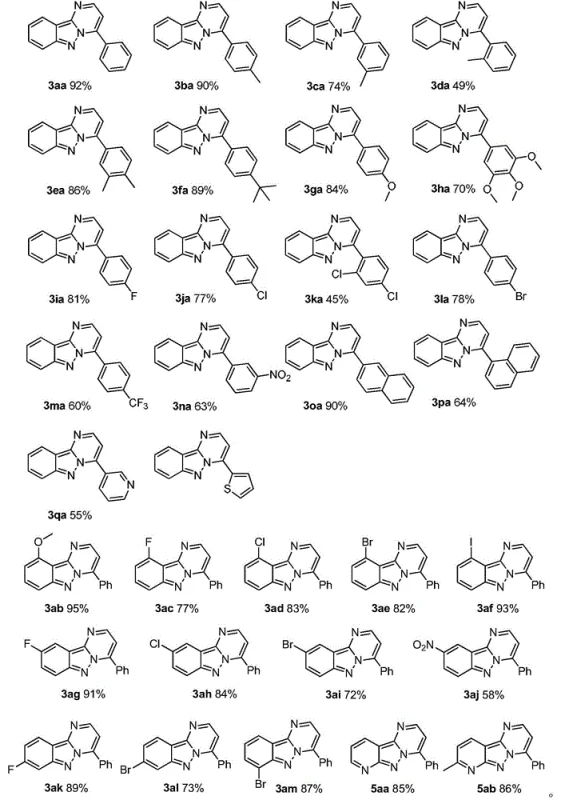
Q: What types of substituents are tolerated on the aromatic aldehyde and indazole rings?
A: The method demonstrates excellent functional group tolerance, accommodating electron-donating groups (methyl, methoxy, tert-butyl) and electron-withdrawing groups (fluorine, chlorine, bromine, nitro, trifluoromethyl) on both the aldehyde and the indazole core.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Pyrimidoindazole Supplier
At NINGBO INNO PHARMCHEM, we recognize the strategic value of efficient synthetic methodologies in the competitive landscape of pharmaceutical development. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that promising laboratory discoveries can be seamlessly translated into reliable commercial supply. We are equipped with rigorous QC labs and adhere to stringent purity specifications to guarantee that every batch of pyrimidoindazole intermediate meets the highest industry standards. Our commitment to technological excellence allows us to offer customized manufacturing solutions that optimize both cost and quality for our global partners.
We invite you to collaborate with us to leverage this advanced metal-free synthesis for your next project. Contact our technical procurement team today to request a Customized Cost-Saving Analysis tailored to your specific volume requirements. We are ready to provide specific COA data and comprehensive route feasibility assessments to support your supply chain strategy and accelerate your path to market.