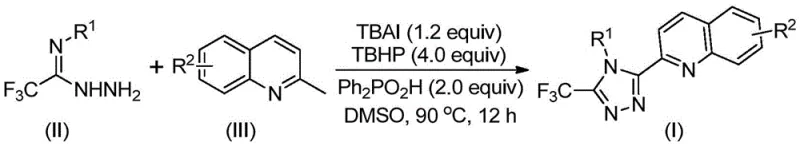
Scalable Synthesis of 3-Quinolyl-5-Trifluoromethyl-1,2,4-Triazoles for Advanced Drug Discovery
The pharmaceutical and fine chemical industries are constantly seeking robust methodologies to construct nitrogen-rich heterocycles, which serve as critical scaffolds in modern drug discovery and material science. Patent CN113307790B introduces a groundbreaking preparation method for 3-quinolyl-5-trifluoromethyl substituted 1,2,4-triazole compounds, addressing significant bottlenecks in traditional synthetic routes. This innovation leverages a metal-free oxidative cyclization strategy that couples readily available 2-methylquinolines with trifluoroacetohydrazides. For R&D directors and procurement specialists, this technology represents a paradigm shift towards more sustainable and cost-effective manufacturing of high-value pharmaceutical intermediates. The ability to introduce both quinoline and trifluoromethyl motifs simultaneously into a triazole core opens new avenues for designing bioactive molecules with enhanced metabolic stability and binding affinity.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of quinolyl-substituted 1,2,4-triazoles has been plagued by inefficiency and operational complexity. Traditional pathways often rely on quinoline-2-carboxylic acid as a starting material, necessitating a tedious five-step reaction sequence to achieve the final heterocyclic structure. This multi-step approach not only results in a dismal overall yield of approximately 17% but also imposes severe reaction conditions that are difficult to control on a large scale. Furthermore, the reliance on multiple isolation and purification steps between each transformation significantly increases solvent consumption and waste generation, driving up the cost of goods sold (COGS). For supply chain managers, these legacy methods pose a substantial risk due to their low throughput and sensitivity to process variations, making them unsuitable for the reliable production of high-purity pharmaceutical intermediates required for clinical trials and commercial launch.
The Novel Approach
In stark contrast, the methodology disclosed in CN113307790B streamlines the synthesis into a direct, one-pot oxidative cyclization. By utilizing cheap and easily obtainable 2-methylquinoline and trifluoroacetohydrazide as starting materials, the process bypasses the need for pre-functionalized carboxylic acid derivatives. The reaction is promoted by a synergistic system of tetrabutylammonium iodide (TBAI) and tert-butyl hydroperoxide (TBHP), facilitating the direct C-H functionalization of the methyl group. This novel approach not only drastically reduces the number of unit operations but also achieves significantly higher yields, with specific examples demonstrating conversion rates exceeding 90%. The elimination of harsh conditions and the use of benign reagents make this method highly attractive for cost reduction in API manufacturing, offering a scalable solution that aligns with green chemistry principles.
Mechanistic Insights into TBAI/TBHP Promoted Oxidative Cyclization
The core of this technological advancement lies in the intricate radical-mediated mechanism driven by the TBAI/TBHP catalytic system. Initially, the iodide species acts as a radical initiator, interacting with the oxidant to generate reactive iodine radicals that abstract a hydrogen atom from the methyl group of the 2-methylquinoline substrate. This generates a benzylic radical which is subsequently oxidized to the corresponding aldehyde intermediate in situ. This transient aldehyde then undergoes a condensation reaction with the hydrazide moiety of the trifluoroacetohydrazide to form a hydrazone intermediate. The presence of diphenylphosphoric acid plays a crucial role in stabilizing intermediates and facilitating proton transfer steps essential for the cyclization cascade.
Following the formation of the hydrazone, the system undergoes a second oxidative iodination event, leading to an intramolecular electrophilic substitution that closes the triazole ring. The final step involves an aromatization process that expels the iodine species, regenerating the catalyst and yielding the stable 3-quinolyl-5-trifluoromethyl-1,2,4-triazole product. This mechanistic pathway is highly efficient because it avoids the accumulation of stable byproducts that typically hinder traditional cyclizations. Understanding this mechanism allows process chemists to fine-tune reaction parameters such as temperature and stoichiometry to maximize selectivity. The robustness of this radical pathway ensures that even with diverse electronic properties on the aromatic rings, the reaction proceeds smoothly, providing a reliable platform for synthesizing complex heterocyclic compounds.
How to Synthesize 3-Quinolyl-5-Trifluoromethyl-1,2,4-Triazoles Efficiently
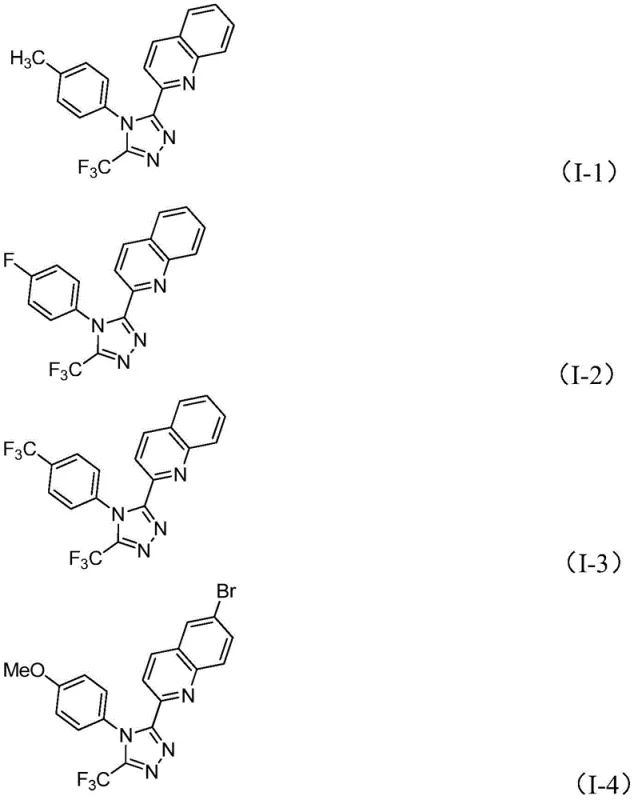
Implementing this synthesis in a laboratory or pilot plant setting requires careful attention to reagent ratios and thermal management, although the procedure itself is remarkably straightforward. The patent outlines a generalized protocol where the key components—TBAI, TBHP, diphenylphosphoric acid, and the two primary substrates—are combined in a polar aprotic solvent like DMSO. The reaction mixture is then heated to a moderate temperature range of 80-100°C, allowing the oxidative cascade to proceed over a period of 8 to 14 hours. This wide operating window provides flexibility for scale-up, ensuring that heat transfer limitations do not compromise reaction completion. The versatility of this method is evidenced by its ability to accommodate various substituents on both the quinoline and the phenyl ring of the hydrazide, as illustrated by the diverse array of successful examples.
- Combine tetrabutylammonium iodide (TBAI), tert-butyl hydroperoxide (TBHP), diphenylphosphoric acid, trifluoroacetohydrazide, and 2-methylquinoline in an organic solvent such as DMSO.
- Heat the reaction mixture to a temperature range of 80-100°C and maintain stirring for 8 to 14 hours to ensure complete oxidative cyclization.
- Upon completion, filter the mixture, adsorb onto silica gel, and purify via column chromatography to isolate the target triazole compound.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this synthetic route offers tangible strategic benefits beyond mere chemical elegance. The shift away from multi-step sequences involving precious metals or hazardous reagents directly translates to a more resilient and cost-efficient supply chain. By simplifying the manufacturing process, companies can reduce their dependency on specialized catalysts that are subject to market volatility and long lead times. Furthermore, the operational simplicity of running reactions under ambient atmospheric conditions removes the capital expenditure associated with maintaining strict inert gas environments, thereby lowering the barrier to entry for contract manufacturing organizations (CMOs) looking to bid on these projects.
- Cost Reduction in Manufacturing: The elimination of heavy metal catalysts is a primary driver for cost optimization in this process. Traditional cross-coupling reactions often require palladium or copper catalysts, which not only carry a high price tag but also necessitate expensive downstream purification steps to meet stringent residual metal limits set by regulatory bodies. By utilizing an organocatalytic system based on inexpensive ammonium salts and peroxides, this method removes the need for scavenger resins or complex extraction protocols. Additionally, the high atom economy and improved yields mean that less raw material is wasted, further driving down the variable costs associated with producing these valuable pharmaceutical intermediates.
- Enhanced Supply Chain Reliability: The starting materials for this synthesis, specifically 2-methylquinolines and trifluoroacetohydrazides, are commodity chemicals that are widely available from multiple global suppliers. This abundance mitigates the risk of supply disruptions that can occur with bespoke or highly specialized reagents. Moreover, the reaction's tolerance to moisture and oxygen means that storage and handling requirements for the reaction mixture are less stringent, reducing the likelihood of batch failures due to environmental exposure. This robustness ensures a consistent flow of materials, which is critical for maintaining production schedules in the fast-paced API manufacturing sector.
- Scalability and Environmental Compliance: From an environmental, health, and safety (EHS) perspective, this method offers significant advantages. The use of DMSO as a solvent, while requiring proper recovery systems, is generally preferred over chlorinated solvents often used in older methodologies. The absence of toxic heavy metals simplifies waste treatment and disposal, aligning with increasingly strict environmental regulations. The process has been demonstrated to work effectively on a gram scale with high conversion, indicating a clear path to kilogram and ton-scale production. This scalability ensures that the technology can support the growing demand for trifluoromethyl-containing drugs without requiring massive re-engineering of existing reactor infrastructure.
Frequently Asked Questions (FAQ)
The following questions address common technical inquiries regarding the implementation and scope of this patented technology. These insights are derived directly from the experimental data and beneficial effects described in the patent documentation, providing clarity for technical teams evaluating this route for potential licensing or development.
Q: Does this synthesis require expensive transition metal catalysts?
A: No, the patented method utilizes a metal-free catalytic system based on tetrabutylammonium iodide (TBAI) and tert-butyl hydroperoxide (TBHP), eliminating the need for costly heavy metals and simplifying downstream purification.
Q: What is the substrate scope for the quinoline component?
A: The method demonstrates excellent tolerance for various substituents on the quinoline ring, including halogens (Br, Cl), alkyl groups (methyl), alkoxy groups (methoxy), and nitro groups at different positions.
Q: Are anhydrous or anaerobic conditions required for this reaction?
A: No, one of the key advantages of this protocol is that it operates effectively under ambient atmospheric conditions without the need for rigorous exclusion of moisture or oxygen, significantly reducing operational complexity.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 3-Quinolyl-5-Trifluoromethyl-1,2,4-Triazole Supplier
At NINGBO INNO PHARMCHEM, we recognize the transformative potential of the synthetic methods described in CN113307790B for the next generation of therapeutic agents. As a leading CDMO partner, we possess the technical expertise to translate these laboratory-scale innovations into robust commercial processes. Our facilities are equipped to handle complex oxidative cyclizations with precision, ensuring that every batch meets the highest standards of quality. We bring extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, leveraging our rigorous QC labs to guarantee stringent purity specifications for every intermediate delivered. Our commitment to process excellence ensures that your supply chain remains uninterrupted and compliant with global regulatory standards.
We invite you to collaborate with us to explore the full capabilities of this metal-free triazole synthesis. By partnering with our technical team, you can gain access to a Customized Cost-Saving Analysis tailored to your specific project needs. We encourage you to contact our technical procurement team today to request specific COA data and route feasibility assessments. Let us help you accelerate your drug development timeline with a reliable supply of high-quality heterocyclic building blocks designed for the future of medicine.