Advanced Palladium-Catalyzed Synthesis of 2-Trifluoromethyl Quinazolinones for Pharmaceutical Manufacturing
Introduction to Next-Generation Quinazolinone Synthesis
The pharmaceutical industry continuously seeks robust methodologies for constructing nitrogen-containing heterocycles, particularly quinazolinones, which serve as privileged scaffolds in numerous bioactive compounds ranging from antifungal agents to anticancer drugs. Patent CN113045503B discloses a groundbreaking preparation method for 2-trifluoromethyl substituted quinazolinone compounds, addressing critical limitations in current synthetic strategies. The introduction of a trifluoromethyl group into these heterocyclic systems is strategically vital, as it significantly enhances physicochemical properties such as electronegativity, metabolic stability, and lipophilicity, thereby improving the overall bioavailability of the parent drug molecule. This innovation represents a paradigm shift from traditional cyclization reactions, offering a streamlined pathway that utilizes transition metal palladium catalysis to achieve high reaction efficiency and broad substrate compatibility.
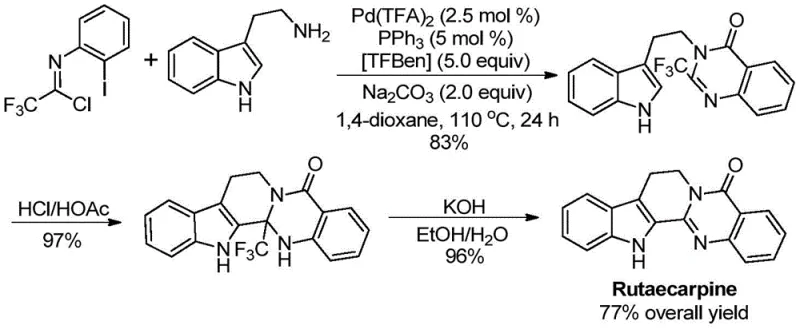
The significance of this technology extends beyond mere academic interest; it provides a tangible solution for the reliable pharmaceutical intermediate supplier seeking to optimize their manufacturing portfolios. By leveraging cheap and easily obtainable starting materials, specifically trifluoroethylimidoyl chloride and various amines, this process eliminates the dependency on costly and unstable reagents often required in legacy syntheses. The method's versatility allows for the design and synthesis of diverse substituted trifluoromethyl quinazolinone compounds, enabling medicinal chemists to rapidly explore structure-activity relationships (SAR) without being hindered by synthetic bottlenecks. Furthermore, the successful application of this method in the efficient synthesis of the complex drug molecule Rutaecarpine underscores its practical utility in real-world drug development scenarios.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of 2-trifluoromethyl-substituted quinazolinone compounds has been plagued by significant technical hurdles that impede efficient large-scale production. Conventional literature reports typically rely on four main strategies, each fraught with distinct disadvantages that impact cost reduction in pharmaceutical intermediate manufacturing. The first common approach involves the cyclization of anthranilamide with ethyl trifluoroacetate, trifluoroacetic anhydride, or trifluoroacetic acid, which often necessitates harsh reaction conditions and generates substantial acidic waste. Another prevalent method utilizes anthranilates reacting with unstable trifluoroacetamides, presenting severe handling challenges and safety risks due to the instability of the intermediates involved in the process.
Additionally, the cyclization of isatoic anhydride with trifluoroacetic anhydride is frequently employed, yet this route suffers from the high cost of the anhydride reagent and limited atom economy. Perhaps the most modern yet still problematic alternative is the T3P-promoted cascade reaction of anthranilic acid, trifluoroacetic acid, and amines; while effective in some contexts, it is generally limited by narrow substrate ranges and the requirement for expensive coupling agents that complicate downstream purification. These collective limitations result in lower overall yields, increased operational complexity, and a higher environmental footprint, making them less attractive for the commercial scale-up of complex polymer additives or fine chemicals where efficiency is paramount.
The Novel Approach
In stark contrast to these legacy methods, the novel palladium-catalyzed carbonylation cascade reaction described in the patent offers a superior alternative that fundamentally reshapes the synthetic landscape for these valuable heterocycles. This innovative approach employs trifluoroethylimidoyl chloride and amines as the primary building blocks, both of which are commercially available, inexpensive, and stable under standard storage conditions. The reaction proceeds through a sophisticated catalytic cycle involving palladium trifluoroacetate and a phosphine ligand, utilizing TFBen (1,3,5-tricarboxylic acid phenol ester) as a safe and solid carbon monoxide surrogate, thereby avoiding the hazards associated with handling gaseous CO directly.
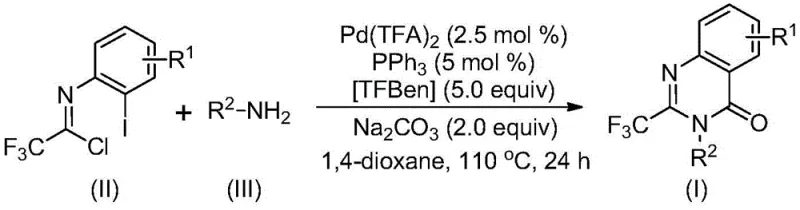
The operational simplicity of this new method cannot be overstated; it requires only mixing the reagents in a common organic solvent like 1,4-dioxane and heating at 110°C for 16 to 30 hours. This mild thermal profile stands in direct opposition to the extreme conditions often demanded by traditional cyclizations. Moreover, the method exhibits exceptional functional group tolerance, accommodating a wide array of substituents on both the aromatic ring and the amine nitrogen, including alkyl, halogen, and trifluoromethyl groups. This broad compatibility ensures that the synthesis of high-purity OLED material precursors or specialized agrochemical intermediates can be achieved with minimal need for protecting group strategies, drastically simplifying the overall synthetic route.
Mechanistic Insights into Pd-Catalyzed Carbonylation Cascade
Understanding the mechanistic underpinnings of this transformation is crucial for R&D directors aiming to optimize the process for specific substrate classes. The reaction is believed to initiate with a base-promoted intermolecular carbon-nitrogen bond coupling between the trifluoroethylimidoyl chloride and the amine, generating a trifluoroacetamidine derivative in situ. Subsequently, the palladium catalyst, generated from palladium trifluoroacetate and triphenylphosphine, undergoes oxidative addition into the carbon-iodine bond of the aromatic ring, forming a key divalent palladium intermediate. This step is critical as it activates the aryl ring for the subsequent carbonylation event, setting the stage for the construction of the quinazolinone core.
Under the heated reaction conditions, the additive TFBen decomposes to release carbon monoxide, which then inserts into the carbon-palladium bond to form an acyl palladium intermediate. This carbonyl insertion is the defining step that introduces the ketone functionality essential for the lactam ring formation. Following this, the base facilitates an intramolecular nucleophilic attack by the nitrogen atom on the acyl palladium species, promoting the formation of a seven-membered ring palladium intermediate. Finally, reductive elimination occurs to release the final 2-trifluoromethyl-substituted quinazolinone compound and regenerate the active palladium catalyst, completing the cycle. This elegant mechanism ensures high atom efficiency and minimizes the formation of side products, contributing to the observed high yields.
From an impurity control perspective, the choice of sodium carbonate as the base and the specific ratio of catalyst to ligand play pivotal roles in suppressing unwanted side reactions such as homocoupling or hydrolysis of the imidoyl chloride. The use of aprotic solvents like 1,4-dioxane further enhances reaction efficiency by effectively dissolving all reactants while stabilizing the charged intermediates involved in the catalytic cycle. This level of mechanistic control allows for the reproducible synthesis of complex molecules with stringent purity specifications, a requirement that is non-negotiable for any reliable electronic chemical supplier or pharmaceutical partner.
How to Synthesize 2-Trifluoromethyl Quinazolinones Efficiently
The execution of this synthesis protocol is designed to be straightforward and adaptable to standard laboratory and pilot plant equipment, ensuring that the transition from discovery to production is seamless. The process begins with the precise weighing of palladium trifluoroacetate, triphenylphosphine, TFBen, sodium carbonate, the specific trifluoroethylimidoyl chloride derivative, and the chosen amine substrate. These components are introduced into a reaction vessel, typically a Schlenk tube for small scale or a stirred tank reactor for larger batches, along with an appropriate volume of 1,4-dioxane to ensure complete dissolution of the solid reagents. The mixture is then subjected to vigorous stirring to create a homogeneous suspension before heating is applied.
- Combine palladium trifluoroacetate, triphenylphosphine, TFBen, sodium carbonate, trifluoroethylimidoyl chloride, and amine in an organic solvent such as 1,4-dioxane.
- Heat the reaction mixture to 110°C and stir for 16 to 30 hours to allow the carbonylation cascade reaction to proceed to completion.
- Perform post-treatment including filtration and silica gel mixing, followed by column chromatography purification to isolate the final 2-trifluoromethyl-substituted quinazolinone compound.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this novel synthetic route offers compelling strategic advantages that directly impact the bottom line and operational resilience. The primary driver for cost reduction in pharmaceutical intermediate manufacturing lies in the raw material selection; by utilizing trifluoroethylimidoyl chlorides and simple amines instead of expensive coupling agents like T3P or unstable anhydrides, the direct material costs are significantly lowered. Furthermore, the elimination of hazardous gaseous carbon monoxide cylinders in favor of the solid CO surrogate TFBen reduces safety compliance costs and simplifies logistics, as solid reagents are far easier to store and transport than compressed gases.
Enhanced supply chain reliability is another critical benefit derived from this technology. The starting materials are widely available from multiple global chemical suppliers, reducing the risk of single-source dependency that often plagues specialty chemical manufacturing. The robustness of the reaction conditions, which tolerate a wide range of functional groups and moisture levels better than traditional methods, means that batch-to-batch variability is minimized. This consistency translates to fewer failed batches and more predictable delivery schedules, allowing supply chain planners to maintain leaner inventory levels while ensuring continuous availability of high-purity API intermediates for downstream formulation.
Scalability and environmental compliance are also markedly improved with this process. The reaction operates in a single pot with a simple workup procedure involving filtration and standard chromatography, which avoids the generation of complex aqueous waste streams associated with multi-step protection-deprotection sequences. The ability to scale this reaction from milligram to gram levels without loss of efficiency demonstrates its potential for ton-scale production, meeting the demands of commercial scale-up of complex pharmaceutical intermediates. Additionally, the high atom economy and reduced use of toxic reagents align with green chemistry principles, facilitating easier regulatory approval and reducing the environmental footprint of the manufacturing facility.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this palladium-catalyzed synthesis method. These insights are derived directly from the experimental data and beneficial effects outlined in the patent documentation, providing clarity on how this technology can be integrated into existing workflows. Understanding these details is essential for technical teams evaluating the feasibility of adopting this route for their specific product pipelines.
Q: What are the primary advantages of this Pd-catalyzed method over traditional cyclization routes?
A: Unlike conventional methods that rely on harsh conditions, expensive pre-activated substrates like trifluoroacetic anhydride, or coupling agents like T3P which often result in low yields and narrow substrate scope, this novel approach utilizes cheap and readily available trifluoroethylimidoyl chlorides and amines. The reaction operates under milder conditions with high efficiency and excellent functional group tolerance, significantly simplifying the synthetic workflow for complex heterocycles.
Q: Can this synthesis method be scaled for industrial production of API intermediates?
A: Yes, the patent explicitly demonstrates that the method is operationally simple and can be extended to the gram level and beyond, providing significant convenience for industrial and medium-scale production applications. The use of standard organic solvents like 1,4-dioxane and commercially available catalysts ensures that the process is robust enough for commercial scale-up of complex pharmaceutical intermediates without requiring exotic equipment.
Q: How does this technology support the synthesis of bioactive molecules like Rutaecarpine?
A: This methodology has been successfully applied to the high-yield total synthesis of the drug molecule Rutaecarpine, achieving an overall yield of 77% across three steps. The ability to construct the quinazolinone core efficiently while tolerating sensitive functional groups makes it an ideal strategy for producing high-purity API intermediates and natural product derivatives with improved metabolic stability due to the trifluoromethyl substitution.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 2-Trifluoromethyl Quinazolinone Supplier
At NINGBO INNO PHARMCHEM, we recognize the transformative potential of advanced catalytic methodologies like the one described in CN113045503B for accelerating drug discovery and development. As a leading CDMO expert, we possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that our clients receive consistent quality regardless of order size. Our state-of-the-art facilities are equipped with rigorous QC labs capable of verifying stringent purity specifications, guaranteeing that every batch of 2-trifluoromethyl quinazolinone meets the highest international standards for pharmaceutical applications.
We invite you to collaborate with us to leverage this cutting-edge technology for your next project. Our technical procurement team is ready to provide a Customized Cost-Saving Analysis tailored to your specific volume requirements, demonstrating exactly how this efficient route can optimize your budget. Please contact us today to request specific COA data for our catalog compounds or to discuss route feasibility assessments for your proprietary molecules, and let us help you bring your innovative therapies to market faster and more economically.