Advanced One-Pot Synthesis of 2-Trifluoromethyl Quinazolinones for Scalable Pharmaceutical Production
Introduction to Next-Generation Quinazolinone Synthesis
The landscape of pharmaceutical intermediate manufacturing is constantly evolving, driven by the need for safer, more efficient, and cost-effective synthetic routes. A pivotal advancement in this domain is detailed in patent CN112480015B, which discloses a robust multi-component one-pot method for synthesizing 2-trifluoromethyl substituted quinazolinones. These heterocyclic scaffolds are not merely academic curiosities; they form the core structure of numerous bioactive molecules with profound therapeutic potential. As illustrated in the structural diversity of known drugs, quinazolinones exhibit a wide spectrum of biological activities, including antifungal, antibacterial, antiviral, anti-inflammatory, and anticancer properties. The introduction of a trifluoromethyl group further enhances these profiles by improving metabolic stability, lipophilicity, and bioavailability, making them highly desirable targets for modern drug discovery programs.
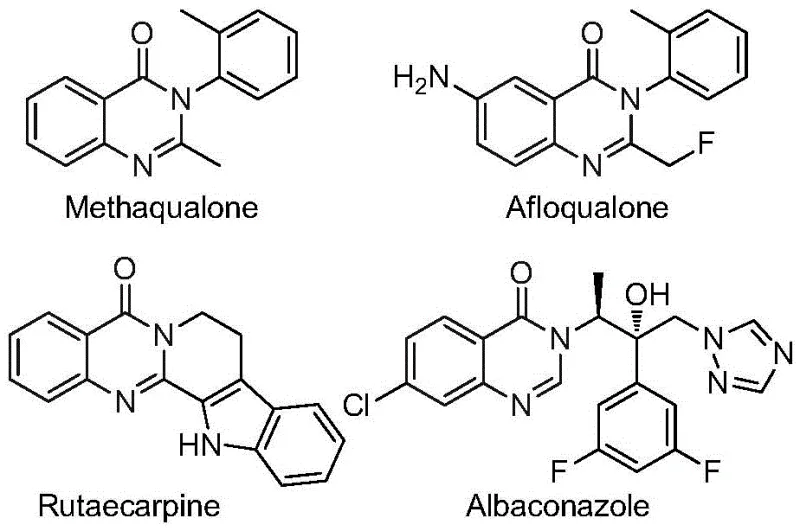
For R&D directors and process chemists, the ability to access these complex fluorinated heterocycles efficiently is paramount. Traditional methods often suffer from significant limitations, such as the requirement for hazardous high-pressure carbon monoxide or the use of expensive, pre-functionalized starting materials. The methodology described in CN112480015B addresses these pain points directly by leveraging a palladium-catalyzed carbonylation cascade. This approach utilizes readily available nitro compounds and trifluoroethylimidoyl chlorides, streamlining the supply chain and reducing the overall environmental footprint of the synthesis. By enabling the construction of the quinazolinone core in a single operational step, this technology represents a significant leap forward in process intensification for fine chemical manufacturing.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of quinazolinone derivatives has relied on several established pathways, each carrying distinct disadvantages that hinder large-scale adoption. Common strategies include ruthenium or platinum-catalyzed reductive N-heterocyclization of nitro-substituted benzamides, which notoriously require high-pressure carbon monoxide conditions. Handling high-pressure CO gas introduces severe safety risks and necessitates specialized, expensive reactor equipment, creating a barrier for many manufacturing facilities. Other methods involve iron-catalyzed condensation reactions or palladium-catalyzed cyclizations using 2-bromoformylaniline or acid anhydrides. These routes often demand substrates that are either costly to procure or require multiple synthetic steps to prepare (pre-activation), thereby increasing the step count and reducing the overall atom economy. Furthermore, many of these conventional protocols exhibit narrow substrate scope, failing to tolerate sensitive functional groups, which limits their utility in the late-stage functionalization of complex drug candidates.
The Novel Approach
In stark contrast, the novel method disclosed in the patent offers a streamlined, one-pot solution that circumvents these traditional bottlenecks. The core innovation lies in the use of nitro compounds as both the nitrogen source and the precursor for the amine functionality via in-situ reduction, coupled with trifluoroethylimidoyl chloride as the electrophilic partner. Crucially, the reaction employs molybdenum hexacarbonyl [Mo(CO)6] as a solid, easy-to-handle carbon monoxide surrogate, eliminating the need for dangerous high-pressure gas cylinders. The reaction proceeds under relatively mild thermal conditions at 120°C in 1,4-dioxane, utilizing a palladium catalyst system composed of PdCl2 and the dppp ligand. This strategy not only simplifies the operational procedure but also dramatically broadens the scope of accessible derivatives. As shown in the general reaction scheme below, the transformation efficiently couples diverse nitroarenes with imidoyl chlorides to yield the target 2-trifluoromethyl quinazolinones with high efficacy.
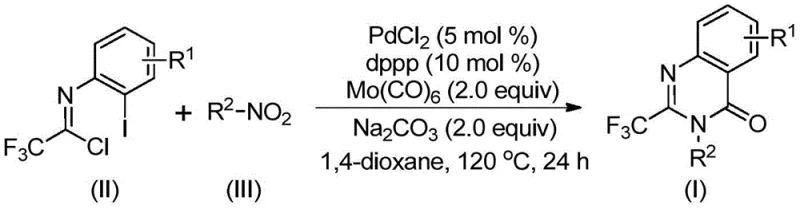
The versatility of this approach is evidenced by its compatibility with a wide array of substituents. Whether the nitro compound bears electron-withdrawing groups like halogens and trifluoromethyl groups or electron-donating alkyl chains, the reaction maintains high efficiency. This flexibility is critical for medicinal chemists who need to rapidly generate libraries of analogs for structure-activity relationship (SAR) studies. By consolidating multiple transformation steps—reduction, coupling, and cyclization—into a single vessel, this method significantly reduces solvent consumption, waste generation, and processing time, aligning perfectly with the principles of green chemistry and sustainable manufacturing.
Mechanistic Insights into Pd-Catalyzed Carbonylation Cascade
Understanding the mechanistic underpinnings of this transformation is essential for optimizing the process and ensuring reproducibility at scale. The reaction is believed to proceed through a sophisticated palladium-catalyzed cascade sequence. Initially, the molybdenum hexacarbonyl serves a dual purpose: it acts as the source of carbon monoxide and potentially facilitates the reduction of the nitro group to the corresponding amine under the reaction conditions. Once the amine is generated in situ, it undergoes a base-promoted nucleophilic attack on the trifluoroethylimidoyl chloride. This intermolecular coupling forms a trifluoroacetamidine intermediate, setting the stage for the cyclization event. Subsequently, the palladium catalyst, activated by the dppp ligand, inserts into the carbon-iodine bond of the aromatic ring (derived from the imidoyl chloride precursor structure), forming a reactive organopalladium species.
As the temperature is maintained at 120°C, the Mo(CO)6 releases carbon monoxide, which then inserts into the carbon-palladium bond to generate an acyl-palladium intermediate. This key step introduces the carbonyl functionality required for the lactam ring formation. Under the influence of the base (sodium carbonate), an intramolecular nucleophilic attack occurs, promoting the formation of a palladium-nitrogen bond and closing the seven-membered cyclic palladium intermediate. The final step involves reductive elimination, which releases the desired 2-trifluoromethyl substituted quinazolinone product and regenerates the active palladium catalyst to continue the cycle. This intricate dance of coordination chemistry ensures high selectivity and minimizes the formation of side products, resulting in the clean impurity profiles observed in the experimental data.
How to Synthesize 2-Trifluoromethyl Quinazolinone Efficiently
Implementing this synthesis in a laboratory or pilot plant setting requires strict adherence to the optimized parameters identified in the patent data. The process is designed to be user-friendly, utilizing standard Schlenk techniques or sealed pressure vessels capable of withstanding the reaction temperature. The stoichiometry is carefully balanced, typically employing a slight excess of the nitro compound (1.2 equivalents) relative to the trifluoroethylimidoyl chloride to drive the reaction to completion. The catalyst loading is kept economical, with PdCl2 used at 5 mol% and the dppp ligand at 10 mol%. For those seeking a standardized protocol to replicate these high-yielding results, the detailed step-by-step instructions are provided below.
- Combine palladium chloride, dppp ligand, sodium carbonate, Mo(CO)6, trifluoroethylimidoyl chloride, and nitro compound in 1,4-dioxane.
- Heat the reaction mixture to 120°C and stir for 16 to 30 hours under inert atmosphere.
- Filter the mixture, mix with silica gel, and purify via column chromatography to isolate the target quinazolinone.
Commercial Advantages for Procurement and Supply Chain Teams
From a commercial perspective, this synthetic route offers compelling advantages that resonate deeply with procurement managers and supply chain leaders. The primary driver for cost reduction lies in the raw material selection. Nitro compounds are commodity chemicals, widely available from global suppliers at a fraction of the cost of specialized boronic acids or pre-activated halides often used in cross-coupling reactions. Furthermore, the elimination of high-pressure carbon monoxide gas removes the need for specialized infrastructure and safety audits associated with toxic gas handling, leading to substantial overhead savings. The use of a solid CO source (Mo(CO)6) simplifies logistics, storage, and handling, reducing the risk of supply chain disruptions caused by hazardous material transport regulations.
- Cost Reduction in Manufacturing: The economic benefits of this process are multifaceted. By avoiding expensive pre-functionalized substrates and utilizing a one-pot methodology, the number of isolation and purification steps is drastically reduced. Each skipped work-up step translates to lower labor costs, reduced solvent usage, and decreased waste disposal fees. Additionally, the reaction demonstrates high atom economy, meaning a larger proportion of the starting mass ends up in the final product. While specific percentage savings depend on local utility costs, the qualitative shift towards cheaper, bulk-available reagents ensures a significantly lower Cost of Goods Sold (COGS) compared to traditional multi-step syntheses.
- Enhanced Supply Chain Reliability: Supply chain resilience is bolstered by the use of robust, commercially mature reagents. Palladium chloride, dppp, and sodium carbonate are standard inventory items for most chemical manufacturers, minimizing the risk of single-source dependency. The reaction's tolerance to various functional groups means that even if a specific substituted nitro compound is temporarily unavailable, alternative analogs can often be sourced or synthesized easily without redesigning the entire process. This flexibility allows procurement teams to maintain continuity of supply even in volatile market conditions, ensuring that production schedules for critical pharmaceutical intermediates remain uninterrupted.
- Scalability and Environmental Compliance: Scaling this reaction from gram to kilogram or ton scale is straightforward due to the absence of hazardous gases and the use of common organic solvents like 1,4-dioxane. The simplified post-treatment process, involving filtration and column chromatography, is easily adaptable to industrial purification methods such as crystallization or preparative HPLC. Moreover, the reduced waste profile aligns with increasingly stringent environmental regulations. By minimizing the generation of heavy metal waste (through efficient catalyst usage) and avoiding toxic gas emissions, this method supports corporate sustainability goals and facilitates smoother regulatory approvals for new drug applications.
Frequently Asked Questions (FAQ)
To assist technical teams in evaluating this technology for their specific applications, we have compiled answers to common questions regarding the process parameters and scope. These insights are derived directly from the experimental data and technical specifications outlined in the patent documentation. Understanding these nuances is crucial for successful technology transfer and process validation.
Q: What are the key advantages of this synthesis method over traditional routes?
A: This method avoids high-pressure carbon monoxide gas and expensive pre-activated substrates, utilizing cheap nitro compounds and solid Mo(CO)6 instead, which significantly enhances operational safety and cost-efficiency.
Q: What is the substrate compatibility of this palladium-catalyzed reaction?
A: The protocol demonstrates excellent functional group tolerance, successfully accommodating various substituents such as halogens (F, Cl, Br), alkyl groups, and trifluoromethyl groups on both the nitro compound and the imidoyl chloride.
Q: Is this process suitable for large-scale industrial production?
A: Yes, the use of commercially available reagents, mild reaction conditions (120°C), and a simple one-pot procedure makes this method highly scalable for commercial manufacturing of pharmaceutical intermediates.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 2-Trifluoromethyl Quinazolinone Supplier
The development of efficient synthetic routes like the one described in CN112480015B underscores the importance of having a knowledgeable manufacturing partner. NINGBO INNO PHARMCHEM stands at the forefront of custom synthesis, possessing extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production. Our team of expert chemists is well-versed in palladium-catalyzed transformations and fluent in optimizing reaction conditions to meet stringent purity specifications required by the global pharmaceutical industry. We understand that every molecule tells a story, and our rigorous QC labs ensure that every batch of 2-trifluoromethyl quinazolinone delivered meets the highest standards of quality and consistency.
We invite you to leverage our technical expertise to accelerate your drug development timeline. Whether you require custom synthesis of novel analogs or reliable supply of key intermediates, our team is ready to provide a Customized Cost-Saving Analysis tailored to your project needs. We encourage potential partners to contact our technical procurement team to request specific COA data and route feasibility assessments. Let us collaborate to bring your next breakthrough therapy to market faster and more efficiently.