Advanced One-Pot Synthesis of 2-Trifluoromethyl Quinazolinones for Commercial API Production
Advanced One-Pot Synthesis of 2-Trifluoromethyl Quinazolinones for Commercial API Production
The pharmaceutical industry continuously seeks robust synthetic routes for heterocyclic scaffolds that possess high metabolic stability and bioavailability. Patent CN112480015B introduces a groundbreaking multi-component one-pot method for synthesizing 2-trifluoromethyl substituted quinazolinones, a privileged structure found in numerous bioactive molecules. This technology addresses critical bottlenecks in traditional heterocycle synthesis by leveraging a palladium-catalyzed carbonylation cascade that operates under relatively mild conditions. The introduction of the trifluoromethyl group is particularly strategic, as fluorine atoms significantly enhance the lipophilicity and metabolic resistance of drug candidates, making this methodology highly relevant for modern drug discovery pipelines. As a leading entity in fine chemical manufacturing, understanding such innovations is crucial for maintaining a competitive edge in the supply of high-purity pharmaceutical intermediates.
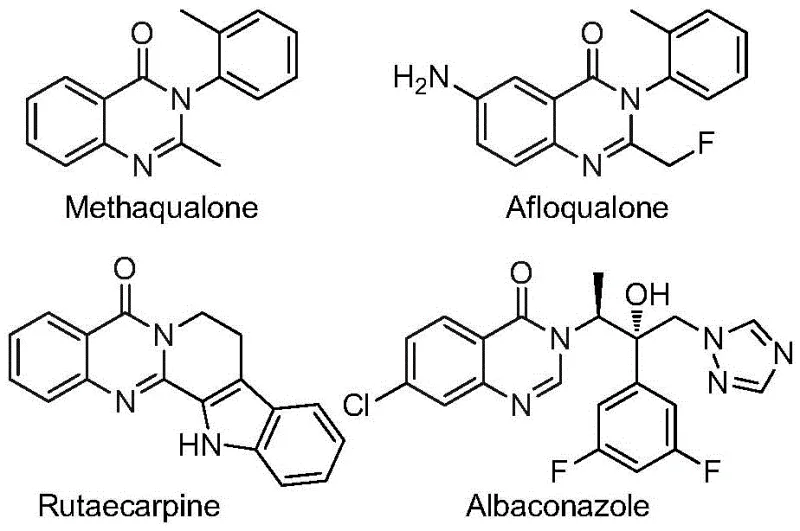
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the construction of the quinazolinone core has relied on methodologies that are fraught with operational hazards and economic inefficiencies. Traditional routes often necessitate the use of high-pressure carbon monoxide gas, which requires specialized autoclaves and rigorous safety protocols that inflate capital expenditure. Furthermore, many established protocols depend on ruthenium or platinum catalysts, which are not only prohibitively expensive but also pose significant challenges regarding residual metal removal in final API products. Other methods involve the use of pre-activated substrates such as acid anhydrides or 2-bromoformylanilines, which add unnecessary synthetic steps and generate substantial chemical waste. These factors collectively result in low atom economy, narrow substrate scope, and yields that are often insufficient for cost-effective commercial manufacturing, thereby limiting the accessibility of these valuable heterocycles for broad therapeutic applications.
The Novel Approach
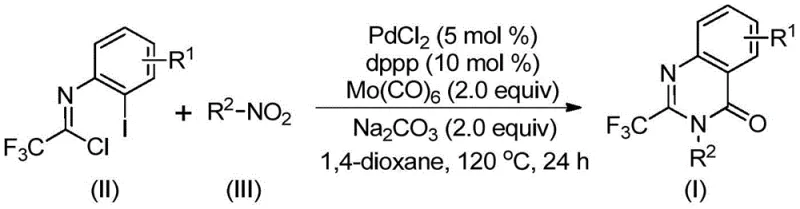
In stark contrast, the methodology disclosed in CN112480015B utilizes a transformative one-pot strategy that dramatically simplifies the synthetic landscape. By employing cheap and readily available nitro compounds alongside trifluoroethylimidoyl chloride, this process bypasses the need for pre-reduced amines or activated acylating agents. The reaction leverages molybdenum hexacarbonyl as a safe, solid carbon monoxide surrogate, effectively eliminating the risks associated with high-pressure gas handling. The system demonstrates exceptional functional group tolerance, accommodating various substituents on both the aromatic ring and the nitrogen atom without compromising efficiency. As illustrated in the specific examples below, this approach consistently delivers high yields across a diverse range of substrates, proving its versatility for generating complex libraries of fluorinated heterocycles essential for medicinal chemistry optimization.
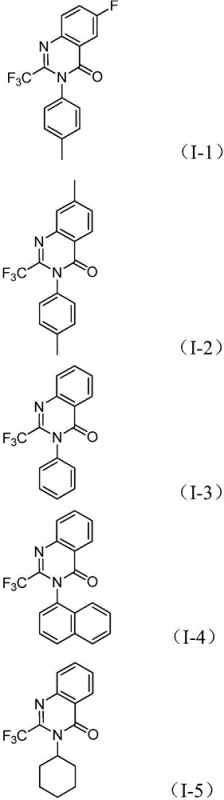
Mechanistic Insights into Palladium-Catalyzed Carbonylation Cascade
The elegance of this synthesis lies in its intricate yet efficient catalytic cycle, which orchestrates multiple bond-forming events in a single vessel. The reaction initiates with the reduction of the nitro group to an amine by molybdenum hexacarbonyl, a critical step that generates the nucleophile in situ. Subsequently, a base-promoted intermolecular coupling occurs between the newly formed amine and the trifluoroethylimidoyl chloride, yielding a trifluoroacetamidine intermediate. The palladium catalyst then inserts into the carbon-iodine bond of the aryl iodide moiety, forming a reactive divalent palladium species. Simultaneously, thermal decomposition of the molybdenum complex releases carbon monoxide, which inserts into the carbon-palladium bond to create an acyl-palladium intermediate. This sequence culminates in the formation of a seven-membered palladacycle, followed by reductive elimination to release the final 2-trifluoromethyl quinazolinone product and regenerate the active catalyst.
Beyond mere bond formation, the mechanistic pathway offers inherent advantages for impurity control, a paramount concern for R&D directors overseeing process development. The use of a specific ligand system, comprising palladium chloride and 1,3-bis(diphenylphosphino)propane (dppp), ensures high regioselectivity and minimizes side reactions such as homocoupling or incomplete carbonylation. The basic conditions provided by sodium carbonate facilitate the cyclization step while neutralizing acidic byproducts, driving the equilibrium towards the desired heterocycle. Furthermore, the one-pot nature of the reaction reduces the exposure of reactive intermediates to the external environment, thereby limiting degradation pathways. This controlled environment results in a cleaner crude reaction profile, which significantly eases the burden on downstream purification processes and ensures that the final material meets stringent purity specifications required for pharmaceutical grade intermediates.
How to Synthesize 2-Trifluoromethyl Quinazolinone Efficiently
Implementing this synthesis requires precise adherence to the optimized reaction parameters to maximize yield and reproducibility. The process involves charging a reaction vessel with the palladium catalyst, ligand, base, and carbon monoxide source before introducing the organic substrates in a suitable aprotic solvent. Maintaining the temperature at 120°C is critical to activate the molybdenum carbonyl complex and drive the carbonylation forward. Detailed standard operating procedures regarding stoichiometry, addition order, and workup protocols are essential for successful technology transfer from lab to pilot plant. For a comprehensive guide on the exact experimental steps and safety precautions, please refer to the standardized synthesis instructions provided below.
- Combine palladium chloride, dppp ligand, sodium carbonate, Mo(CO)6, trifluoroethylimidoyl chloride, and nitro compound in an organic solvent like 1,4-dioxane.
- Heat the reaction mixture to 120°C and maintain stirring for 16 to 30 hours to ensure complete conversion.
- Upon completion, filter the mixture, mix with silica gel, and purify via column chromatography to isolate the target quinazolinone.
Commercial Advantages for Procurement and Supply Chain Teams
From a procurement and supply chain perspective, this patented methodology offers substantial strategic benefits that translate directly into operational resilience and cost efficiency. By shifting away from hazardous high-pressure gases and precious metal catalysts like ruthenium, manufacturers can significantly reduce both capital investment in specialized equipment and ongoing operational expenditures. The reliance on commodity chemicals such as nitro compounds and simple aryl iodides ensures a stable and diversified supply base, mitigating the risks associated with sourcing exotic or single-source reagents. Moreover, the simplified workup procedure, which avoids complex extraction sequences, reduces solvent consumption and waste disposal costs, aligning with modern green chemistry initiatives and environmental compliance standards.
- Cost Reduction in Manufacturing: The elimination of high-pressure carbon monoxide infrastructure removes a major barrier to entry and reduces facility maintenance costs significantly. Additionally, the substitution of expensive ruthenium or platinum catalysts with a more economical palladium system, combined with the use of cheap nitro starting materials, drastically lowers the bill of materials. The high reaction efficiency and yield minimize raw material waste, ensuring that every kilogram of input translates effectively into saleable product, thereby optimizing the overall cost of goods sold for API manufacturing.
- Enhanced Supply Chain Reliability: The starting materials, specifically nitro compounds and trifluoroethylimidoyl chlorides, are widely available from global chemical suppliers, reducing lead times and dependency on niche vendors. The robustness of the reaction conditions allows for flexible scheduling and batch processing without the need for highly specialized operator training or rare equipment availability. This reliability ensures consistent production timelines, enabling supply chain managers to meet demanding delivery schedules for downstream pharmaceutical clients without unexpected interruptions caused by reagent shortages or equipment failures.
- Scalability and Environmental Compliance: The one-pot nature of the reaction inherently reduces the number of unit operations, making the process easier to scale from gram to multi-kilogram quantities without losing efficiency. The use of 1,4-dioxane as a solvent, while requiring careful management, is a well-understood industrial solvent with established recovery protocols. By avoiding the generation of heavy metal waste associated with stoichiometric reductants and minimizing solvent swaps, the process facilitates easier regulatory approval and reduces the environmental footprint, supporting sustainable manufacturing goals.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis technology. These insights are derived directly from the experimental data and beneficial effects described in the patent documentation, providing clarity on feasibility and performance. Understanding these aspects is vital for stakeholders evaluating the integration of this method into their existing production workflows or R&D pipelines.
Q: What are the advantages of using nitro compounds over traditional amines in this synthesis?
A: Nitro compounds are significantly cheaper and more readily available than pre-activated amines or acid anhydrides. This method utilizes them directly, bypassing expensive pre-functionalization steps and reducing overall raw material costs.
Q: Does this process require high-pressure carbon monoxide equipment?
A: No, the process utilizes Mo(CO)6 as a solid carbon monoxide substitute. This eliminates the need for dangerous high-pressure gas cylinders and specialized autoclaves, greatly simplifying safety protocols and equipment requirements.
Q: Is this method suitable for large-scale industrial production?
A: Yes, the patent explicitly states the method is operable at the gram level and designed for scalability. The use of common solvents like dioxane and robust catalytic systems supports commercial scale-up for API manufacturing.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 2-Trifluoromethyl Quinazolinone Supplier
At NINGBO INNO PHARMCHEM, we recognize the transformative potential of advanced catalytic methodologies like the one described in CN112480015B for accelerating drug development. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that promising laboratory discoveries can be seamlessly transitioned into reliable industrial supply. We operate stringent purity specifications and maintain rigorous QC labs equipped with state-of-the-art analytical instrumentation to guarantee that every batch of 2-trifluoromethyl quinazolinone meets the highest quality standards required by global regulatory bodies.
We invite you to collaborate with us to leverage this efficient synthesis route for your specific project needs. Our technical procurement team is ready to provide a Customized Cost-Saving Analysis tailored to your volume requirements, demonstrating how this technology can optimize your budget. Please contact us today to request specific COA data and route feasibility assessments, and let us partner with you to secure a stable, high-quality supply of these critical pharmaceutical intermediates.