Advanced Palladium-Catalyzed Synthesis of 3-Arylquinolin-2(1H)-one Derivatives for Commercial Scale-up
The pharmaceutical and fine chemical industries are constantly seeking robust, scalable methodologies for constructing privileged heterocyclic scaffolds, particularly those found in bioactive small molecules. A significant breakthrough in this domain is detailed in Chinese Patent CN113045489B, which discloses a novel preparation method for 3-arylquinolin-2(1H)-one derivatives. This specific class of compounds serves as a critical structural motif in a vast array of therapeutic agents, ranging from antibiotics and antiplatelet drugs to potent antitumor agents and receptor antagonists. The strategic importance of this scaffold is underscored by its presence in complex drug candidates such as MAP Kinase inhibitors and HBV inhibitors, highlighting the urgent need for efficient synthetic access.
This patented technology represents a paradigm shift in how these valuable intermediates are manufactured. By leveraging a palladium-catalyzed aminocarbonylation strategy, the process transforms simple, commercially available starting materials into high-value heterocycles with remarkable efficiency. For R&D directors and procurement specialists alike, understanding the nuances of this invention is crucial, as it offers a pathway to reduce production costs while enhancing the purity profile of the final active pharmaceutical ingredients (APIs). The method not only simplifies the operational workflow but also aligns with modern green chemistry principles by minimizing waste and avoiding hazardous reagents often associated with classical heterocycle synthesis.
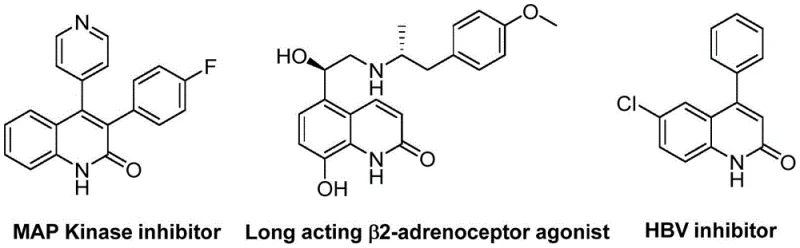
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the construction of the quinolin-2(1H)-one core has relied on classical named reactions such as the Vilsmeier-Haack, Knorr, and Friedlander condensations. While these methods are well-documented in academic literature, they suffer from significant drawbacks when applied to large-scale industrial manufacturing. Traditional approaches often necessitate the use of corrosive reagents, extreme temperatures, and multi-step sequences that degrade overall yield. Furthermore, the functional group tolerance in these legacy methods is frequently poor, requiring extensive protecting group strategies that add unnecessary complexity and cost to the supply chain. The reliance on stoichiometric amounts of toxic reagents also poses substantial environmental and safety challenges, complicating waste disposal and regulatory compliance for commercial facilities.
The Novel Approach
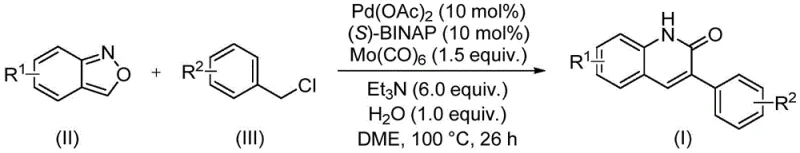
In stark contrast, the methodology described in patent CN113045489B introduces a streamlined, transition-metal-catalyzed solution that elegantly bypasses these historical bottlenecks. The core innovation involves the use of benzisoxazole as a unique dual-purpose reagent, functioning simultaneously as the nitrogen source and the formyl source for the ring construction. This clever design eliminates the need for external carbon monoxide gas or dangerous formylating agents, significantly enhancing process safety. The reaction proceeds under relatively mild conditions using a palladium catalyst system, allowing for the direct coupling of benzisoxazoles with benzyl chlorides. This one-pot transformation not only reduces the number of unit operations but also dramatically improves the atom economy of the process, making it an ideal candidate for cost-effective commercial scale-up.
Mechanistic Insights into Palladium-Catalyzed Aminocarbonylation
The success of this synthetic route hinges on a sophisticated catalytic cycle driven by a palladium complex coordinated with chiral phosphine ligands. Specifically, the system utilizes palladium acetate in conjunction with (S)-1,1'-binaphthyl-2,2'-bis-diphenylphosphine ((S)-BINAP) and molybdenum hexacarbonyl (Mo(CO)6). In this mechanism, Mo(CO)6 serves as a safe, solid surrogate for carbon monoxide, releasing CO in situ under the reaction conditions to facilitate the carbonylation step. The benzisoxazole substrate undergoes oxidative addition and subsequent ring-opening events mediated by the palladium center, effectively unmasking the reactive nitrogen and carbonyl equivalents needed for cyclization. This intricate interplay between the metal catalyst and the heterocyclic precursor ensures high regioselectivity and minimizes the formation of unwanted byproducts.
From an impurity control perspective, this mechanism offers distinct advantages over acid-catalyzed condensations. The mild basic conditions provided by triethylamine prevent the degradation of acid-sensitive functional groups, which is a common issue in traditional Friedlander syntheses. Moreover, the specific ligand environment created by (S)-BINAP helps to stabilize the active catalytic species, preventing the aggregation of palladium black and ensuring consistent reaction kinetics throughout the 26-hour reaction window. The result is a clean reaction profile where the desired 3-arylquinolin-2(1H)-one is formed with high fidelity, simplifying downstream purification and ensuring that the final product meets stringent pharmaceutical purity specifications without the need for exhaustive recrystallization.
How to Synthesize 3-Arylquinolin-2(1H)-one Efficiently
To implement this cutting-edge synthesis in a laboratory or pilot plant setting, operators must adhere to precise stoichiometric ratios and thermal parameters to maximize yield. The process begins with the careful charging of the catalytic components and substrates into a pressure-rated vessel, followed by the addition of the solvent system. Maintaining the correct temperature profile is critical, as deviations can impact the rate of CO release from the molybdenum source and the stability of the palladium catalyst. The following guide outlines the standardized operational procedure derived from the patent examples to ensure reproducible results.
- Combine palladium acetate, (S)-BINAP, molybdenum carbonyl, triethylamine, water, benzisoxazole, and benzyl chloride in a sealed tube with DME solvent.
- Heat the reaction mixture to 100°C and maintain stirring for approximately 26 hours to ensure complete conversion.
- Upon completion, filter the mixture, mix with silica gel, and purify via column chromatography to isolate the target 3-arylquinolin-2(1H)-one derivative.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this patented methodology translates into tangible strategic benefits that extend beyond mere chemical yield. The shift towards this catalytic protocol addresses several critical pain points associated with the sourcing and manufacturing of complex heterocyclic intermediates. By simplifying the synthetic route and utilizing commodity chemicals, manufacturers can achieve significant cost reductions while simultaneously de-risking the supply chain against raw material volatility. The robustness of the process also ensures greater reliability in meeting delivery schedules, a key metric for maintaining continuity in API production lines.
- Cost Reduction in Manufacturing: The economic viability of this process is driven by the use of inexpensive, readily available starting materials such as benzyl chlorides and substituted benzisoxazoles. Unlike traditional methods that may require custom-synthesized precursors or expensive gaseous reagents, this protocol leverages bulk chemicals that are accessible from multiple global suppliers. Furthermore, the elimination of multiple synthetic steps and protecting group manipulations drastically reduces the consumption of solvents and reagents, leading to substantial savings in raw material costs and waste disposal fees. The high efficiency of the catalyst system also means that lower loading levels can be utilized without compromising conversion, further optimizing the cost structure.
- Enhanced Supply Chain Reliability: Supply chain resilience is significantly bolstered by the broad substrate scope of this reaction. Because the method tolerates a wide variety of functional groups, including halogens, alkoxy groups, and nitriles, manufacturers are not locked into a single, rigid synthetic path. This flexibility allows for the rapid adaptation of the process to accommodate alternative starting materials should supply disruptions occur. Additionally, the use of stable, solid reagents like Mo(CO)6 instead of hazardous gases simplifies logistics and storage requirements, reducing the regulatory burden and ensuring smoother transportation and handling across international borders.
- Scalability and Environmental Compliance: Scaling this reaction from gram to kilogram quantities is facilitated by its operation in standard organic solvents like ethylene glycol dimethyl ether (DME) at moderate temperatures. The absence of highly exothermic steps or dangerous high-pressure gas inputs makes the process inherently safer for large-scale reactors. From an environmental standpoint, the improved atom economy and the avoidance of stoichiometric toxic waste streams align perfectly with increasingly strict global environmental regulations. This compliance reduces the risk of production shutdowns due to environmental audits and positions the manufacturing site as a sustainable partner for eco-conscious pharmaceutical clients.
Frequently Asked Questions (FAQ)
The following questions address common technical inquiries regarding the implementation and scope of this synthesis technology. These answers are derived directly from the experimental data and claims within the patent documentation to provide accurate guidance for process development teams evaluating this route for their specific projects.
Q: What is the primary innovation in this synthesis method compared to traditional routes?
A: The primary innovation lies in the utilization of benzisoxazole as a dual-function reagent, serving simultaneously as both the nitrogen source and the formyl source. This eliminates the need for separate formylation steps and harsh reagents typically required in Vilsmeier-Haack or Friedlander reactions, thereby streamlining the synthetic pathway and improving atom economy.
Q: What are the optimal reaction conditions for maximizing yield in this protocol?
A: The patent specifies that optimal results are achieved using a catalytic system of palladium acetate and (S)-BINAP with molybdenum hexacarbonyl as the CO source. The reaction is conducted in ethylene glycol dimethyl ether (DME) at a temperature of 100°C for a duration of 26 hours, utilizing triethylamine as the base.
Q: Does this method support a wide range of functional groups on the substrate?
A: Yes, the methodology demonstrates excellent functional group tolerance. It successfully accommodates various substituents including halogens (Cl, F), alkoxy groups (methoxy), alkyl groups (tert-butyl), and electron-withdrawing groups (cyano, trifluoromethyl) on both the benzisoxazole and benzyl chloride components, allowing for the diverse synthesis of quinolinone derivatives.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 3-Arylquinolin-2(1H)-one Supplier
The technological potential of this palladium-catalyzed aminocarbonylation route is immense, offering a clear path to high-quality pharmaceutical intermediates. At NINGBO INNO PHARMCHEM, we possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your transition from lab bench to market is seamless. Our facility is equipped with rigorous QC labs and adheres to stringent purity specifications, guaranteeing that every batch of 3-arylquinolin-2(1H)-one derivative meets the exacting standards required for drug substance manufacturing.
We invite you to leverage our technical expertise to optimize your supply chain. Contact our technical procurement team today to request a Customized Cost-Saving Analysis tailored to your specific volume requirements. We are ready to provide specific COA data and comprehensive route feasibility assessments to demonstrate how this innovative synthesis can drive value for your organization.