Scalable Metal-Free Synthesis of 5-Trifluoromethyl-1,2,3-Triazoles for Pharmaceutical Applications
Scalable Metal-Free Synthesis of 5-Trifluoromethyl-1,2,3-Triazoles for Pharmaceutical Applications
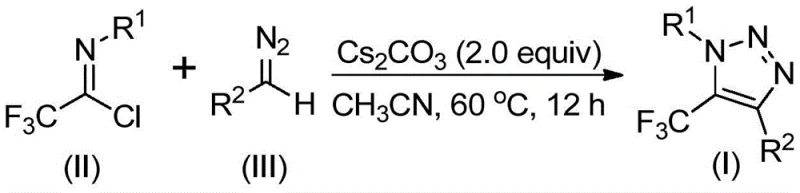
The development of efficient synthetic routes for fluorinated heterocycles remains a critical priority in modern medicinal chemistry, particularly for the construction of robust molecular scaffolds found in bioactive agents. Patent CN113121462B introduces a groundbreaking preparation method for 5-trifluoromethyl substituted 1,2,3-triazole compounds, addressing long-standing safety and efficiency challenges in the industry. This technology leverages a base-promoted cyclization strategy that completely eliminates the need for transition metal catalysts and hazardous organic azides, which have traditionally plagued this chemical space. By utilizing readily available trifluoroethylimidoyl chloride and diazo compounds as starting materials, the process offers a streamlined pathway to high-value intermediates essential for pharmaceutical and agrochemical development. For R&D directors and procurement specialists, this innovation represents a significant opportunity to optimize supply chains while adhering to stricter environmental and safety regulations.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of trifluoromethyl-substituted 1,2,3-triazoles has relied heavily on two primary methodologies, both of which present substantial drawbacks for large-scale manufacturing. The first approach involves copper-catalyzed [3+2] cycloaddition reactions between alkynes and organic azides, followed by trifluoromethylation. This route is problematic due to the inherent instability and toxicity of organic azides, posing severe explosion risks during handling and storage. Furthermore, the use of copper catalysts necessitates rigorous downstream purification steps to remove trace metal residues, which is a critical requirement for pharmaceutical intermediates intended for human consumption. The second conventional method employs organocatalytic 1,3-dipolar cycloaddition involving azides and trifluoromethyl ketones, which similarly suffers from the safety hazards associated with azide reagents. These legacy processes often result in complex impurity profiles and increased operational costs related to safety containment and waste disposal.
The Novel Approach
In stark contrast to these hazardous legacy methods, the novel approach detailed in the patent utilizes a metal-free, base-promoted reaction between trifluoroethylimidoyl chloride and diazo compounds. This strategy fundamentally shifts the risk profile by replacing explosive azides with more stable diazo precursors and avoiding heavy metal contamination entirely. The reaction proceeds through a mechanism where the base facilitates an intermolecular nucleophilic addition-elimination process, followed by an intramolecular 5-endo-dig cyclization to form the triazole ring. This not only simplifies the reaction setup but also drastically reduces the complexity of the workup procedure. As illustrated in the reaction scheme below, the transformation is direct and atom-economical, providing a cleaner route to the desired 5-trifluoromethyl-1,2,3-triazole core structure.
The visual comparison of synthetic strategies highlights the elegance of the new method (c) compared to traditional copper-catalyzed or organocatalytic routes (a and b). By bypassing the formation of triazolide metal intermediates and avoiding toxic azide reagents, this new protocol aligns perfectly with the principles of green chemistry. For a reliable pharmaceutical intermediate supplier, adopting such a methodology ensures a more consistent supply of high-purity materials while minimizing the environmental footprint associated with heavy metal waste and hazardous reagent disposal.
Mechanistic Insights into Base-Promoted Cyclization
The mechanistic pathway of this transformation is driven by the basicity of cesium carbonate, which activates the diazo compound for nucleophilic attack on the electrophilic carbon of the trifluoroethylimidoyl chloride. This initial step forms a key intermediate that undergoes elimination of chloride, promoting the formation of a new carbon-carbon bond. Subsequently, the nitrogen atom of the diazo moiety attacks the nitrile carbon in a 5-endo-dig cyclization fashion, closing the triazole ring. This specific cyclization mode is favored under the mild thermal conditions provided (50-70°C), ensuring high regioselectivity for the 5-trifluoromethyl isomer. The absence of transition metals means that the reaction coordinate is purely governed by electronic and steric factors of the organic substrates, allowing for predictable outcomes across a wide range of substituents.
From an impurity control perspective, this mechanism offers distinct advantages. Since no metal catalyst is involved, there is no risk of metal-ligand complexation side reactions that often lead to difficult-to-remove byproducts. The primary byproducts are inorganic salts derived from the base and the leaving group, which are easily removed during the aqueous workup or filtration steps. This results in a crude product with a significantly cleaner impurity profile, reducing the burden on purification teams. For quality control laboratories, this translates to faster release times and higher confidence in the consistency of the final API intermediate. The robustness of this mechanism against various functional groups, such as esters, ketones, and phosphonates, further underscores its utility in diversifying chemical libraries for drug discovery programs.
How to Synthesize 5-Trifluoromethyl-1,2,3-Triazoles Efficiently
The operational simplicity of this synthesis makes it highly attractive for both laboratory-scale optimization and pilot plant production. The standard protocol involves charging a reactor with cesium carbonate, the specific trifluoroethylimidoyl chloride, and the corresponding diazo compound in an aprotic solvent such as acetonitrile. The mixture is then heated to a moderate temperature, typically around 60°C, and stirred for a period of 8 to 16 hours to ensure full conversion. Following the reaction, the mixture is filtered to remove inorganic salts, and the filtrate is concentrated. The resulting crude material can be purified via standard column chromatography or recrystallization techniques to yield the target triazole compound with high purity. Detailed standardized synthesis steps are provided in the guide below.
- Mix cesium carbonate, trifluoroethylimidoyl chloride, and diazo compound in an aprotic solvent like acetonitrile.
- Heat the reaction mixture to 50-70°C and stir for 8-16 hours to ensure complete conversion.
- Filter the mixture, concentrate, and purify the crude product via column chromatography to obtain the target triazole.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this patented synthesis route offers compelling economic and logistical benefits that extend beyond simple yield metrics. The elimination of expensive transition metal catalysts, such as copper, directly reduces the raw material cost per kilogram of the final product. Moreover, the removal of metal catalysts obviates the need for specialized scavenging resins or complex extraction protocols designed to lower metal content to ppm levels, thereby simplifying the manufacturing workflow and reducing processing time. The use of commercially available and stable starting materials ensures a reliable supply chain, mitigating the risks associated with sourcing hazardous or custom-synthesized reagents that often face regulatory shipping restrictions.
- Cost Reduction in Manufacturing: The economic impact of switching to this metal-free process is substantial. By removing the dependency on precious or transition metal catalysts, manufacturers can achieve significant cost savings on reagent procurement. Additionally, the simplified workup procedure reduces the consumption of solvents and purification media, leading to lower overall production costs. The avoidance of hazardous azides also lowers insurance and safety compliance costs, contributing to a more favorable cost structure for high-purity pharmaceutical intermediates.
- Enhanced Supply Chain Reliability: Supply chain continuity is greatly improved by the use of robust, shelf-stable reagents. Unlike organic azides, which may require special permits for transport and storage, the diazo compounds and imidoyl chlorides used in this process are more manageable within standard chemical logistics frameworks. This stability allows for larger batch sizes and longer inventory holding periods without degradation, ensuring that production schedules are met consistently without unexpected delays caused by reagent instability or regulatory hold-ups.
- Scalability and Environmental Compliance: The mild reaction conditions and absence of heavy metals make this process inherently scalable and environmentally friendly. Scaling from gram to kilogram quantities does not require significant re-engineering of the process parameters, as the thermal profile is gentle and easy to control. Furthermore, the reduction in hazardous waste generation aligns with increasingly stringent global environmental regulations, facilitating easier permitting for manufacturing facilities and enhancing the sustainability profile of the final chemical products.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis technology. These answers are derived directly from the experimental data and technical specifications outlined in the patent documentation, providing clarity on safety, scalability, and product quality. Understanding these details is crucial for technical teams evaluating the feasibility of integrating this route into their existing manufacturing portfolios.
Q: Why is this new synthesis method safer than traditional triazole production?
A: Traditional methods often rely on organic azides, which are toxic and potentially explosive, or copper catalysts that require complex removal. This patented route utilizes stable diazo compounds and imidoyl chlorides under metal-free conditions, significantly enhancing operational safety.
Q: What are the optimal reaction conditions for scaling this process?
A: The patent specifies using cesium carbonate (2.0 equivalents) in acetonitrile at 60°C for 12 hours. These mild conditions facilitate easy scale-up without requiring high-pressure equipment or cryogenic temperatures.
Q: Can this method accommodate diverse functional groups on the triazole ring?
A: Yes, the method demonstrates broad substrate scope, tolerating various substituents on both the N-1 and C-4 positions, including esters, ketones, phosphonates, and halogenated aryl groups, making it versatile for drug discovery.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 5-Trifluoromethyl-1,2,3-Triazole Supplier
At NINGBO INNO PHARMCHEM, we recognize the transformative potential of this metal-free synthesis technology for the production of advanced pharmaceutical intermediates. As a dedicated CDMO partner, we possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your project transitions smoothly from benchtop discovery to full-scale manufacturing. Our state-of-the-art facilities are equipped with rigorous QC labs capable of meeting stringent purity specifications, guaranteeing that every batch of 5-trifluoromethyl-1,2,3-triazole delivered meets the highest industry standards for potency and impurity control.
We invite you to collaborate with us to leverage this innovative chemistry for your next drug development program. Our technical procurement team is ready to provide a Customized Cost-Saving Analysis tailored to your specific volume requirements. Please contact us today to request specific COA data and route feasibility assessments, and let us demonstrate how our expertise can accelerate your timeline to market while optimizing your production costs.