Scalable Metal-Free Synthesis of Trifluoromethyl Triazoles for Advanced Pharmaceutical Intermediates
Introduction to Advanced Triazole Manufacturing Technologies
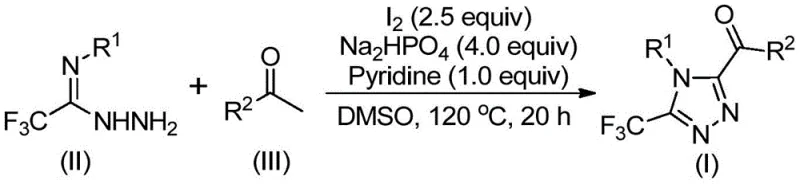
The pharmaceutical and fine chemical industries are constantly seeking robust synthetic routes for nitrogen-containing heterocycles, particularly those incorporating trifluoromethyl groups which enhance metabolic stability and lipophilicity. A significant breakthrough in this domain is detailed in patent CN113105402B, which discloses a highly efficient preparation method for 3,4,5-trisubstituted 1,2,4-triazole compounds. This technology addresses critical pain points in modern organic synthesis by eliminating the reliance on toxic heavy metal catalysts and stringent anhydrous conditions. For R&D directors and procurement managers alike, this represents a paradigm shift towards greener, more cost-effective manufacturing of key pharmaceutical intermediates used in drugs like Maraviroc and Sitagliptin. The method leverages a unique iodine-promoted mechanism that ensures high purity and excellent functional group tolerance, making it an ideal candidate for reliable pharmaceutical intermediate supplier networks aiming to optimize their supply chains.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Traditionally, the synthesis of polysubstituted 1,2,4-triazoles has often relied on transition metal catalysis or harsh reaction conditions that pose significant challenges for industrial scale-up. Conventional pathways frequently necessitate the use of expensive palladium or copper catalysts, which not only inflate raw material costs but also introduce complex downstream purification steps to remove trace metal residues to meet stringent regulatory standards for active pharmaceutical ingredients. Furthermore, many existing protocols require strictly anhydrous and oxygen-free environments, demanding specialized equipment and increasing operational complexity. These factors collectively contribute to longer lead times and higher production costs, creating bottlenecks for manufacturers striving for efficiency in high-purity pharmaceutical intermediate production. The presence of heavy metals can also limit the application of these intermediates in sensitive biological assays or final drug formulations.
The Novel Approach
In stark contrast, the methodology outlined in the patent introduces a metal-free strategy utilizing elemental iodine and dimethyl sulfoxide (DMSO) to drive the reaction forward under relatively mild conditions. This innovative approach capitalizes on the Kornblum oxidation capability of the iodine-DMSO system to generate reactive alpha-diketone intermediates in situ, which subsequently undergo tandem cyclization with trifluoroethylimide hydrazides. By avoiding toxic heavy metals and eliminating the need for inert atmosphere protection, this process drastically simplifies the operational workflow and reduces the environmental footprint. The use of cheap, commercially available starting materials such as aryl ethyl ketones further enhances the economic viability of this route. This novel approach offers a streamlined pathway for the commercial scale-up of complex pharmaceutical intermediates, ensuring consistent quality and supply continuity.
Mechanistic Insights into Iodine-Promoted Cyclization
The core of this synthetic innovation lies in the dual role of the iodine-DMSO system, which acts as both an oxidant and a promoter for the cyclization event. Mechanistically, the reaction initiates with the iodination of the aryl ethyl ketone, followed by a Kornblum oxidation to form an aryl alpha-diketone species. This highly reactive intermediate then undergoes a dehydration condensation with the trifluoroethylimide hydrazide to form a hydrazone intermediate. Subsequently, under the influence of the base (sodium dihydrogen phosphate and pyridine) and continued heating, an intramolecular cyclization occurs to close the triazole ring. This cascade sequence is highly efficient and minimizes the formation of side products, thereby simplifying the impurity profile. Understanding this mechanism is crucial for process chemists aiming to replicate these results, as it highlights the importance of precise temperature control during the two distinct heating stages to maximize yield and selectivity.
From an impurity control perspective, the absence of transition metals significantly reduces the risk of metal-catalyzed side reactions or decomposition pathways that often plague traditional methods. The mild basic conditions provided by the phosphate buffer system help maintain the stability of the trifluoromethyl group, preventing defluorination which can occur under strongly acidic or basic conditions. Additionally, the broad substrate scope demonstrated in the patent suggests that electron-donating and electron-withdrawing groups on the aromatic rings are well-tolerated, allowing for the synthesis of a diverse library of analogs without compromising purity. This robustness is essential for R&D teams exploring structure-activity relationships (SAR) where rapid access to varied derivatives is required without the need for extensive method re-optimization for each new substrate.
How to Synthesize 3,4,5-Trisubstituted 1,2,4-Triazole Efficiently
Implementing this synthesis route requires careful attention to the two-stage heating protocol and reagent stoichiometry to ensure optimal conversion. The process begins by dissolving the aryl ethyl ketone and a portion of elemental iodine in DMSO, followed by an initial heating period to generate the oxidized intermediate. Once this stage is complete, the remaining reagents including the hydrazide, base, and additional iodine are introduced to drive the cyclization. The detailed standardized synthesis steps, including specific molar ratios and workup procedures, are critical for achieving the reported yields and purity levels. For a comprehensive guide on executing this protocol in a laboratory or pilot plant setting, please refer to the structured instructions below.
- Combine aryl ethyl ketone and elemental iodine in dimethyl sulfoxide (DMSO) and heat the mixture to 90-110°C for 4-6 hours to initiate Kornblum oxidation.
- Add additional iodine, sodium dihydrogen phosphate, pyridine, and trifluoroethylimide hydrazide to the reaction solution.
- Raise the temperature to 110-130°C and maintain for 12-20 hours to complete the cyclization, followed by filtration and column chromatography purification.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this metal-free synthesis route offers substantial strategic advantages regarding cost structure and supply reliability. By eliminating the dependency on precious metal catalysts, manufacturers can achieve significant cost reduction in pharmaceutical intermediate manufacturing, as the price volatility associated with metals like palladium is completely removed from the equation. Furthermore, the use of commodity chemicals such as aryl ketones and iodine ensures a stable and diversified supply base, reducing the risk of raw material shortages that can disrupt production schedules. The simplified post-treatment process, which avoids complex metal scavenging steps, also translates to reduced processing time and lower utility consumption, contributing to overall operational efficiency.
- Cost Reduction in Manufacturing: The elimination of expensive transition metal catalysts directly lowers the bill of materials, while the simplified purification process reduces solvent usage and waste disposal costs. This economic efficiency allows for more competitive pricing structures without compromising on the quality of the final product, making it an attractive option for high-volume production runs where margin optimization is critical.
- Enhanced Supply Chain Reliability: Since the starting materials are widely available commodity chemicals rather than specialized organometallic reagents, the supply chain is inherently more resilient to market fluctuations. This stability ensures consistent delivery timelines and reduces the lead time for high-purity pharmaceutical intermediates, enabling downstream drug manufacturers to maintain steady production schedules and meet market demand effectively.
- Scalability and Environmental Compliance: The method's compatibility with standard reactor setups and its operation under non-anhydrous conditions facilitate easy scale-up from laboratory to commercial tonnage. Additionally, the absence of heavy metals simplifies wastewater treatment and aligns with increasingly stringent environmental regulations, supporting sustainable manufacturing practices and reducing the regulatory burden on production facilities.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding this synthesis technology, derived from the specific advantages and operational details found in the patent documentation. These insights are intended to clarify the feasibility of implementing this route for various applications and to highlight the key differentiators compared to legacy methods. Understanding these aspects is vital for stakeholders evaluating the potential integration of this technology into their existing manufacturing portfolios.
Q: Does this synthesis method require expensive transition metal catalysts?
A: No, the method described in patent CN113105402B utilizes elemental iodine as a non-metal promoter, completely eliminating the need for costly palladium or copper catalysts and simplifying downstream purification.
Q: What are the critical reaction conditions for optimal yield?
A: The process requires a two-stage heating protocol in DMSO: an initial oxidation phase at 90-110°C followed by a cyclization phase at 110-130°C, ensuring high conversion without anhydrous conditions.
Q: Is this method suitable for large-scale industrial production?
A: Yes, the patent explicitly states the method is easily scalable from gram level to industrial production due to the use of cheap, commercially available raw materials and simple operational procedures.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 3,4,5-Trisubstituted 1,2,4-Triazole Supplier
At NINGBO INNO PHARMCHEM, we recognize the transformative potential of metal-free synthetic methodologies in advancing the production of complex heterocyclic scaffolds. As a leading CDMO partner, we possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that innovative lab-scale discoveries like this iodine-promoted triazole synthesis are successfully translated into robust industrial processes. Our commitment to quality is underpinned by stringent purity specifications and rigorous QC labs that utilize advanced analytical techniques to verify the absence of metal residues and confirm structural integrity, guaranteeing that every batch meets the highest industry standards.
We invite global partners to collaborate with us to leverage this cost-effective and scalable technology for their specific project needs. By engaging with our technical procurement team, you can request a Customized Cost-Saving Analysis tailored to your volume requirements, along with specific COA data and route feasibility assessments. Let us help you optimize your supply chain and accelerate your drug development timeline with our reliable supply of high-quality pharmaceutical intermediates.