Advanced Palladium-Catalyzed Carbonylation for Scalable 2-Trifluoromethyl Imidazole Production
The pharmaceutical and agrochemical industries continuously seek robust synthetic methodologies to access nitrogen-containing heterocycles functionalized with trifluoromethyl groups, driven by the profound impact of the CF3 moiety on metabolic stability, lipophilicity, and bioavailability. As detailed in patent CN111423381B, a groundbreaking preparation method for 2-trifluoromethyl substituted imidazole compounds has been developed, addressing critical bottlenecks in current manufacturing landscapes. This innovation leverages a transition metal palladium-catalyzed carbonylation series reaction, utilizing readily available starting materials to construct complex molecular architectures with high precision. The significance of this technology is underscored by the prevalence of imidazole scaffolds in bioactive molecules, ranging from histamine receptor antagonists like Alcaftadine to sophisticated NHC ligands used in coordination catalysis.
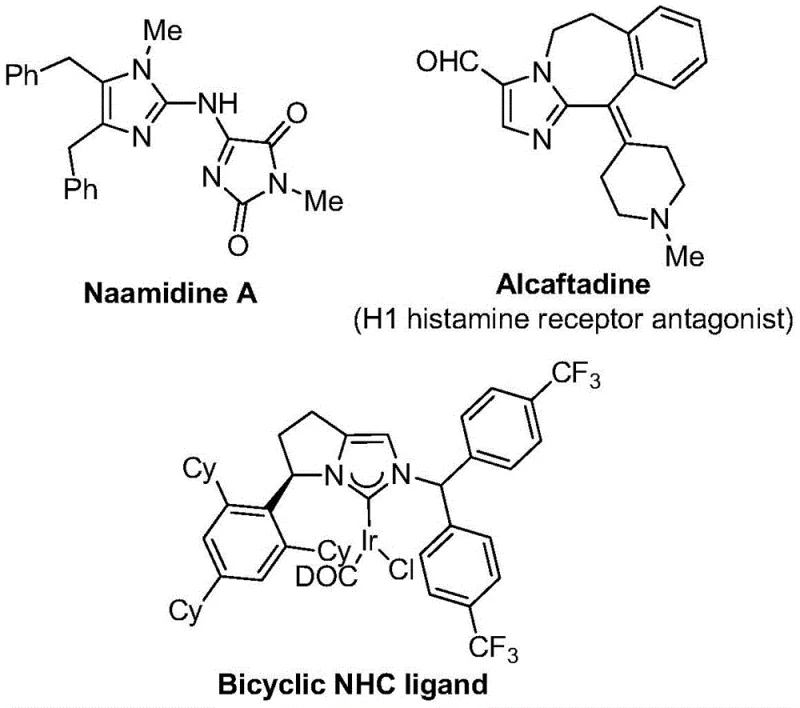
By introducing the trifluoromethyl group directly into the heterocyclic backbone through this novel route, manufacturers can significantly enhance the physicochemical properties of lead compounds without resorting to late-stage fluorination strategies that often suffer from poor selectivity. The method described in the patent represents a paradigm shift towards more sustainable and operationally simple processes, capable of tolerating a wide array of functional groups while maintaining high reaction efficiency. For global supply chain leaders, this translates to a reliable pathway for producing high-purity pharmaceutical intermediates that meet the stringent quality standards required for modern drug development pipelines.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Traditional synthetic routes for accessing trifluoromethyl-substituted nitrogen heterocycles have historically relied on the direct reaction of synthons bearing the trifluoromethyl group with suitable substrates, a strategy fraught with significant operational and safety challenges. Commonly employed trifluoromethyl synthons, such as trifluorodiazoethane, are inherently unstable and pose severe safety risks due to their explosive nature, necessitating specialized equipment and rigorous safety protocols that drive up capital expenditure. Furthermore, alternative reagents like trifluoroethylimidoyl halides have seen limited application due to difficulties in controlling regioselectivity and the requirement for harsh reaction conditions that can degrade sensitive functional groups on the substrate. These conventional methods often result in complex impurity profiles, requiring extensive and costly purification steps that reduce overall process mass intensity and yield. Additionally, the reliance on high-pressure carbon monoxide gas in many carbonylation reactions introduces significant logistical hurdles for large-scale manufacturing, limiting the ability of facilities to scale up production safely and economically.
The Novel Approach
In stark contrast to these legacy techniques, the novel approach disclosed in the patent utilizes a mild, palladium-catalyzed multicomponent coupling reaction that operates under exceptionally gentle conditions, typically at 30°C. This method ingeniously combines trifluoroethylimidoyl chloride, propargylamine, and diaryl iodonium salts in the presence of a formic acid and acetic anhydride mixture, which serves as a safe and controllable carbon monoxide alternative. The reaction scheme illustrates a streamlined one-pot process that constructs the imidazole ring while simultaneously installing the trifluoromethyl and aryl ketone functionalities with remarkable atom economy.
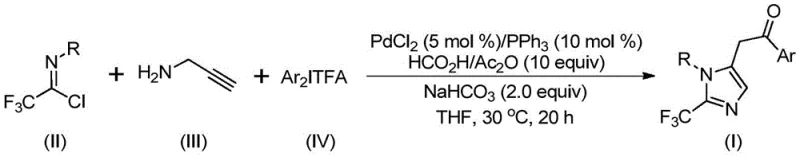
This strategic design eliminates the need for hazardous gaseous CO and unstable diazo compounds, thereby drastically simplifying the engineering controls required for production. The compatibility of this method with various substituted aryl groups on both the imidoyl chloride and the iodonium salt components allows for the rapid generation of diverse chemical libraries, facilitating the optimization of structure-activity relationships in drug discovery. By shifting the synthetic burden to readily available and stable precursors, this approach offers a scalable solution that aligns perfectly with the goals of cost reduction in pharmaceutical intermediate manufacturing.
Mechanistic Insights into Pd-Catalyzed Carbonylative Cyclization
The mechanistic pathway of this transformation is a sophisticated orchestration of organometallic steps that ensures high fidelity in product formation. The process initiates with the formation of an intermolecular carbon-nitrogen bond promoted by the base, yielding a trifluoroacetamidine intermediate which subsequently undergoes isomerization. Concurrently, the palladium catalyst activates the alkyne moiety of the propargylamine through palladation, generating a key alkenyl palladium intermediate. This species then isomerizes to a more stable alkyl palladium intermediate, setting the stage for the crucial carbonylation step. The carbon monoxide required for this insertion is generated in situ from the decomposition of the formic acid and acetic anhydride additive system, which then inserts into the palladium-carbon bond to form an acyl palladium intermediate. The cycle concludes with the oxidative addition of the diaryl iodonium salt to form a transient tetravalent palladium species, followed by reductive elimination to release the final 2-trifluoromethyl-substituted imidazole product and regenerate the active catalyst.
From an impurity control perspective, the mild thermal conditions (30°C) play a pivotal role in suppressing side reactions such as polymerization of the alkyne or decomposition of the sensitive imidoyl chloride. The use of sodium bicarbonate as a mild base further ensures that acid-sensitive functionalities on the aromatic rings remain intact throughout the reaction course. The broad substrate tolerance is evidenced by the successful synthesis of various derivatives, including those with electron-donating groups like methoxy and tert-butyl, as well as electron-withdrawing groups like nitro and halogens, all yielding the target structures with high purity.
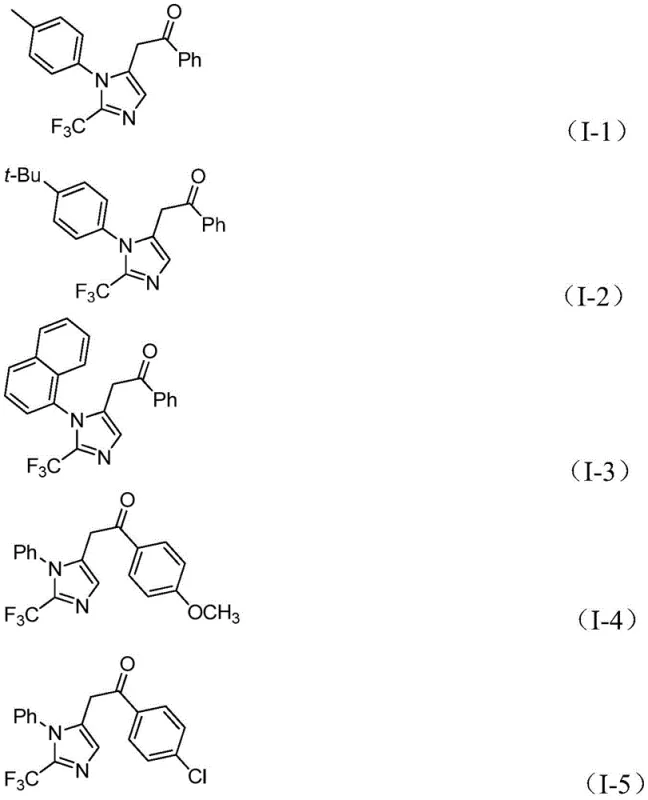
This mechanistic robustness allows for the predictable synthesis of complex analogues, providing R&D teams with the confidence to design diverse molecular libraries without fear of catastrophic reaction failure. The precise control over the catalytic cycle minimizes the formation of palladium black and other metal aggregates, ensuring that the final product meets stringent heavy metal specifications essential for pharmaceutical applications.
How to Synthesize 2-Trifluoromethyl Imidazole Efficiently
The operational simplicity of this synthesis makes it highly attractive for both laboratory-scale discovery and pilot-plant production. The procedure involves charging a reactor with the palladium catalyst system, ligands, and the three primary building blocks in a suitable aprotic solvent like tetrahydrofuran. The reaction is allowed to proceed under ambient pressure, removing the need for autoclaves or high-pressure gas lines. Detailed standard operating procedures for scaling this reaction are critical for maintaining consistency.
- Combine palladium chloride, triphenylphosphine, sodium bicarbonate, acetic anhydride, formic acid, trifluoroethylimidoyl chloride, propargylamine, and diaryl iodonium salt in an organic solvent such as THF.
- Stir the reaction mixture at a mild temperature of 30°C for a duration of 16 to 24 hours to ensure complete conversion.
- Upon completion, filter the mixture, mix with silica gel, and purify via column chromatography to isolate the target 2-trifluoromethyl substituted imidazole compound.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this patented methodology offers transformative advantages in terms of cost structure and supply reliability. The shift away from exotic, high-cost fluorinating agents to commodity chemicals like propargylamine and aromatic amines fundamentally alters the cost basis of the final intermediate. By utilizing a catalyst system based on palladium chloride and triphenylphosphine, which are industry standards with established supply chains, the process avoids the volatility associated with proprietary or single-source reagents. This stability in raw material sourcing is crucial for maintaining uninterrupted production schedules in a global market prone to logistical disruptions.
- Cost Reduction in Manufacturing: The economic benefits of this process are driven primarily by the elimination of expensive and hazardous reagents such as trifluorodiazoethane, which require specialized synthesis and handling infrastructure. By replacing high-pressure carbon monoxide gas with a liquid mixture of formic acid and acetic anhydride, the facility requirements are significantly downgraded, leading to substantial capital expenditure savings and lower operational overheads. Furthermore, the high reaction efficiency and yield reported in the patent examples imply a reduction in solvent usage and waste disposal costs per kilogram of product, contributing to a leaner and more profitable manufacturing model. The ability to run the reaction at near-ambient temperature also results in drastic energy savings compared to processes requiring reflux or cryogenic conditions.
- Enhanced Supply Chain Reliability: The starting materials for this synthesis, including trifluoroethylimidoyl chloride and diaryl iodonium salts, are derived from widely available aromatic amines and boronic acids, ensuring a robust and diversified supply base. This reduces the risk of supply chain bottlenecks that often plague processes dependent on niche fluorine chemistry suppliers. The modular nature of the reaction allows for the easy substitution of different aryl groups, meaning that if a specific precursor faces shortage, the synthetic route can be adapted to use alternative analogues without redesigning the entire process. This flexibility provides a strategic buffer against market volatility and ensures continuous availability of critical intermediates for downstream API production.
- Scalability and Environmental Compliance: The process is inherently scalable, having been demonstrated effectively at the gram level with clear pathways for expansion to multi-kilogram and ton-scale production. The mild reaction conditions minimize the formation of thermal runaway hazards, making the scale-up process safer and more predictable for engineering teams. From an environmental standpoint, the avoidance of toxic gases and the use of a catalytic amount of palladium align with green chemistry principles, simplifying regulatory compliance and waste treatment. The straightforward post-treatment involving filtration and column chromatography facilitates the recovery of solvents and catalysts, further enhancing the sustainability profile of the manufacturing operation.
Frequently Asked Questions (FAQ)
Understanding the technical nuances of this synthesis is vital for stakeholders evaluating its integration into existing production workflows. The following questions address common inquiries regarding the practical implementation and capabilities of this technology, drawing directly from the experimental data and disclosures within the patent documentation.
Q: What serves as the carbon monoxide source in this synthesis?
A: The process utilizes a combination of formic acid and acetic anhydride as an in situ carbon monoxide alternative, eliminating the need for handling hazardous high-pressure CO gas cylinders directly.
Q: What are the optimal reaction conditions for this transformation?
A: The reaction proceeds efficiently at a mild temperature of 30°C over a period of 16 to 24 hours using tetrahydrofuran (THF) as the preferred organic solvent.
Q: Which catalyst system is employed for this carbonylation?
A: The method employs a cost-effective palladium catalyst system consisting of palladium chloride (PdCl2) and triphenylphosphine (PPh3) ligand.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 2-Trifluoromethyl Imidazole Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical role that advanced synthetic methodologies play in accelerating drug development and securing supply chains. Our team of expert chemists possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the transition from laboratory bench to industrial reactor is seamless and efficient. We are committed to delivering high-purity 2-trifluoromethyl imidazole intermediates that adhere to stringent purity specifications, supported by our rigorous QC labs equipped with state-of-the-art analytical instrumentation. Our capability to implement complex palladium-catalyzed carbonylations allows us to offer customized solutions that meet the specific needs of global pharmaceutical partners.
We invite you to collaborate with us to leverage this cutting-edge technology for your next project. Contact our technical procurement team today to request a Customized Cost-Saving Analysis tailored to your volume requirements. We are ready to provide specific COA data and comprehensive route feasibility assessments to demonstrate how our manufacturing expertise can drive value and efficiency in your supply chain.