Scalable Synthesis of Tucatinib Intermediates: A Technical Breakthrough for Commercial Manufacturing
The pharmaceutical industry is constantly seeking robust, scalable pathways for the production of high-value oncology therapeutics, and the recent disclosure in patent CN111825604A offers a transformative approach to synthesizing Tucatinib, a potent HER2 tyrosine kinase inhibitor. This intellectual property details a novel synthetic methodology that fundamentally restructures the manufacturing landscape for this critical API intermediate, moving away from the cumbersome, low-yielding protocols of the past. By leveraging a strategic nucleophilic aromatic substitution using accessible halide salts, the inventors have successfully bypassed the need for exotic catalytic systems that have historically plagued the supply chain. For R&D directors and procurement specialists alike, this represents a significant pivot towards process intensification, where the elimination of precious metal catalysts directly correlates to reduced material costs and simplified regulatory filings. The technical depth of this patent suggests a mature understanding of reaction engineering, particularly in the optimization of base-mediated couplings that were previously thought to require protective group chemistry. As we analyze the implications of CN111825604A, it becomes clear that this is not merely an incremental improvement but a foundational shift enabling reliable tucatinib intermediate supplier capabilities on a global scale.
The Limitations of Conventional Methods vs. The Novel Approach
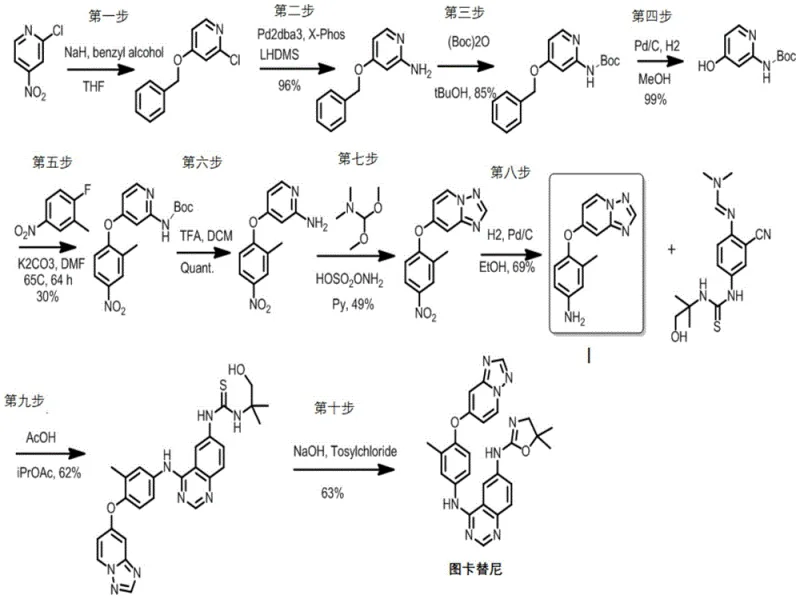
The Limitations of Conventional Methods
Prior to this innovation, the synthesis of Tucatinib and its precursors was heavily reliant on the methodologies described in legacy patents such as WO2007059257A2, which presented substantial bottlenecks for industrial adoption. The conventional route was characterized by an excessive number of synthetic steps, often requiring up to eight distinct transformations to reach the key intermediate, resulting in a cumulative overall yield that was economically unsustainable at approximately 5.7%. Furthermore, these legacy processes depended heavily on the use of sophisticated and expensive transition metal catalysts, specifically Pd2dba3, alongside strong, moisture-sensitive bases like lithium hexamethyldisilazide (LiHDMS). The reliance on such reagents introduces significant supply chain volatility, as the sourcing of high-purity palladium complexes can be inconsistent and cost-prohibitive for large-volume manufacturing. Additionally, the necessity for amino protection strategies to prevent side reactions added further complexity, extending reaction times to over 64 hours for certain intermediates and generating substantial chemical waste. These factors combined to create a high barrier to entry for generic manufacturers and increased the final cost of goods for the active pharmaceutical ingredient, limiting patient access to this life-saving therapy.
The Novel Approach
In stark contrast, the methodology outlined in CN111825604A introduces a streamlined, convergent synthesis that drastically reduces the step count while simultaneously enhancing reaction efficiency and selectivity. The core innovation lies in the direct substitution reaction between a halide salt of Formula VI and a nitro-substituted benzene derivative (Formula VII) under alkaline conditions, which successfully constructs the critical ether linkage in a single operational step. This approach completely obviates the need for the aforementioned palladium catalysts and sensitive lithium bases, replacing them with inexpensive, commodity chemicals such as potassium hydroxide and triethylamine. The patent data indicates that this simplified protocol can achieve yields exceeding 60% for the key intermediate (Formula VIII) with purity levels surpassing 90%, a dramatic improvement over the 30% yield and lower purity associated with the multi-step prior art. By removing the requirement for amino protection and deprotection sequences, the new route not only saves time but also minimizes the generation of hazardous byproducts, aligning perfectly with modern green chemistry principles. This robust chemical design ensures that the production of high-purity pharmaceutical intermediates can be executed with greater predictability and lower capital expenditure.
Mechanistic Insights into Base-Mediated Nucleophilic Aromatic Substitution
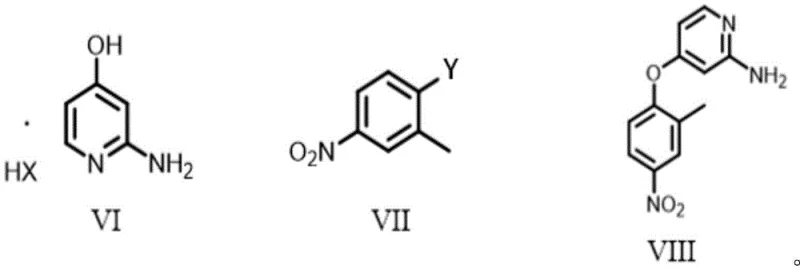
The success of this novel synthetic route hinges on a meticulously optimized nucleophilic aromatic substitution (SNAr) mechanism, where the electronic properties of the reactants are leveraged to drive the reaction forward without external catalytic assistance. In this transformation, the phenolic oxygen of the pyridine derivative (Formula VI), activated by the basic environment, acts as a potent nucleophile attacking the electron-deficient aromatic ring of the fluoronitrobenzene (Formula VII). The presence of the strong electron-withdrawing nitro group ortho or para to the leaving group (fluorine) significantly lowers the energy of the LUMO (Lowest Unoccupied Molecular Orbital) of the aromatic ring, facilitating the addition-elimination sequence typical of SNAr reactions. The patent specifies the use of a dual-base system, often combining an inorganic base like KOH with an organic amine like triethylamine, which serves to both deprotonate the phenol and scavenge the acidic byproduct (HF or HCl), thereby shifting the equilibrium towards product formation. This careful balancing of base strength and solubility prevents the degradation of sensitive functional groups while ensuring complete conversion of the starting materials within a timeframe of less than 18 hours. Such mechanistic precision allows for the direct formation of the diaryl ether bond, which is the structural backbone of the Tucatinib molecule, with minimal formation of regio-isomers or hydrolysis byproducts.
Furthermore, the control of impurities in this process is achieved through the strategic selection of reaction conditions that favor the desired kinetic pathway over thermodynamic side reactions. By maintaining the reaction temperature within a controlled range of 25°C to 70°C and utilizing polar aprotic solvents like DMF or DMSO, the solvation of the nucleophile is enhanced without promoting competing elimination reactions. The patent highlights that the use of the halide salt of Formula VI, rather than the free base alone, provides a more stable and handleable starting material that dissolves readily in the reaction medium, ensuring homogeneous mixing and consistent heat transfer. This homogeneity is crucial for preventing local hot spots that could lead to the decomposition of the nitro group or the formation of tar-like polymeric impurities. Post-reaction workup involves a simple precipitation into ice water followed by recrystallization, a physical purification method that effectively removes inorganic salts and unreacted starting materials, yielding a product with purity levels suitable for subsequent coupling steps without the need for chromatographic purification. This level of impurity control is vital for meeting the stringent specifications required for clinical-grade pharmaceutical intermediates.
How to Synthesize Tucatinib Key Intermediate Efficiently
The practical execution of this synthesis involves a straightforward protocol that begins with the charging of the halide salt of Formula VI and the fluoronitrobenzene derivative into a reactor containing a suitable solvent such as DMF. The reaction is initiated by the addition of the base system, typically potassium hydroxide and triethylamine, under controlled temperature conditions to manage the exotherm and ensure safety. Detailed standardized operating procedures for stoichiometry, addition rates, and quenching protocols are essential for reproducibility, and the full technical guide for these steps is provided below for our manufacturing partners.
- Prepare the reaction mixture by combining the halide salt of Formula VI or its free base with Formula VII in a polar aprotic solvent like DMF.
- Add a dual-base system consisting of Potassium Hydroxide (KOH) and Triethylamine (Et3N) to facilitate the nucleophilic aromatic substitution under mild heating.
- Monitor the reaction via HPLC until starting material is consumed, then precipitate the product in ice water and recrystallize to achieve high purity.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of the synthesis route described in CN111825604A offers compelling economic and logistical benefits that extend far beyond simple yield improvements. The most immediate impact is the drastic reduction in raw material costs, driven by the elimination of high-value palladium catalysts and specialized lithium reagents that are subject to volatile market pricing and geopolitical supply risks. By substituting these with commodity bases like potassium hydroxide, the bill of materials for the intermediate is significantly lowered, allowing for more competitive pricing structures in the final API market. Moreover, the simplification of the synthetic route from eight steps to a far more concise sequence reduces the overall manufacturing cycle time, thereby increasing the throughput capacity of existing production facilities without the need for major capital investment in new equipment. This efficiency gain translates directly into enhanced supply chain reliability, ensuring that critical drug substances can be delivered to patients without the delays often associated with complex, multi-step syntheses.
- Cost Reduction in Manufacturing: The removal of expensive transition metal catalysts such as Pd2dba3 eliminates the need for costly metal scavenging steps and rigorous residual metal testing, which are significant cost drivers in pharmaceutical manufacturing. Additionally, the higher yields achieved in each step mean that less raw material is wasted, improving the overall atom economy and reducing the cost per kilogram of the final product. The use of common solvents and reagents also simplifies waste management and disposal, further contributing to substantial cost savings in the overall production budget.
- Enhanced Supply Chain Reliability: By relying on widely available starting materials like halide salts and nitro-aromatics, the risk of supply disruption due to single-source dependency is minimized. The robustness of the reaction conditions, which do not require inert atmospheres or cryogenic temperatures typical of organolithium chemistry, allows for production in a wider range of manufacturing sites, diversifying the supply base. This flexibility ensures continuity of supply even in the face of regional disruptions, providing a secure source of high-purity pharmaceutical intermediates for downstream formulation.
- Scalability and Environmental Compliance: The process is inherently designed for scale-up, utilizing standard unit operations like filtration and crystallization rather than complex chromatography, which is difficult to implement on a multi-ton scale. The reduction in hazardous reagents and the generation of less toxic waste streams align with increasingly strict environmental regulations, reducing the compliance burden and potential liability for manufacturers. This green chemistry approach not only safeguards the environment but also enhances the corporate sustainability profile of the supply chain partners involved.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this novel Tucatinib synthesis route, derived directly from the experimental data and claims within the patent documentation. These insights are intended to clarify the feasibility of technology transfer and the expected quality parameters for the resulting intermediates. Understanding these nuances is critical for technical teams evaluating the integration of this process into their existing manufacturing portfolios.
Q: How does the new synthesis route improve yield compared to prior art?
A: The patented method achieves a yield of over 60% for the key intermediate (Formula VIII) in a single step, whereas previous methods required six steps with a cumulative yield of only 30%.
Q: Does this process require expensive transition metal catalysts?
A: No, the novel route eliminates the need for costly palladium catalysts (like Pd2dba3) and lithium bases (LiHDMS), utilizing common inorganic bases like KOH instead.
Q: Is this synthesis method suitable for large-scale industrial production?
A: Yes, the process uses readily available raw materials, avoids harsh conditions, and simplifies purification, making it highly suitable for commercial scale-up and GMP manufacturing.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Tucatinib Intermediate Supplier
At NINGBO INNO PHARMCHEM, we recognize that the transition from laboratory patent to commercial reality requires a partner with deep technical expertise and proven manufacturing capabilities. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the promising yields and purity profiles described in CN111825604A can be reliably replicated on an industrial scale. We maintain stringent purity specifications and operate rigorous QC labs equipped with state-of-the-art analytical instrumentation to guarantee that every batch of Tucatinib intermediate meets the highest global regulatory standards. Our commitment to quality assurance means that we can navigate the complexities of process validation and regulatory filing support, providing our clients with a seamless path to market approval.
We invite procurement leaders and R&D directors to engage with our technical procurement team to discuss how this optimized synthesis route can be tailored to your specific volume requirements and cost targets. By requesting a Customized Cost-Saving Analysis, you can gain a clear understanding of the potential economic benefits of switching to this catalytic-free methodology. We encourage you to contact us today to obtain specific COA data and route feasibility assessments, allowing you to make informed decisions that will strengthen your supply chain and enhance your competitive position in the oncology therapeutic market.