Advanced Palladium-Catalyzed Synthesis of 2-Trifluoromethyl Quinazolinone Derivatives for Commercial Scale-Up
Advanced Palladium-Catalyzed Synthesis of 2-Trifluoromethyl Quinazolinone Derivatives for Commercial Scale-Up
The pharmaceutical industry continuously seeks robust synthetic methodologies to access complex heterocyclic scaffolds that serve as the backbone for next-generation therapeutics. Patent CN112125856A introduces a significant advancement in this domain by disclosing a novel preparation method for 2-trifluoromethyl substituted quinazolinone derivatives. These structures are not merely academic curiosities; they are critical pharmacophores found in a wide array of bioactive molecules exhibiting anti-inflammatory, antiviral, antifungal, anticonvulsant, and anticancer properties. The strategic incorporation of the trifluoromethyl group is particularly valued in medicinal chemistry for its ability to enhance metabolic stability, lipophilicity, and bioavailability without significantly increasing steric bulk. As a reliable pharmaceutical intermediate supplier, understanding the nuances of such synthetic breakthroughs is essential for delivering high-purity compounds that meet the rigorous demands of drug discovery pipelines.
The significance of this technology extends beyond simple molecule construction; it addresses fundamental challenges in process chemistry regarding safety and scalability. Quinazolinone derivatives, such as those depicted in the structural diversity of known drugs, require precise functionalization to unlock their full therapeutic potential. 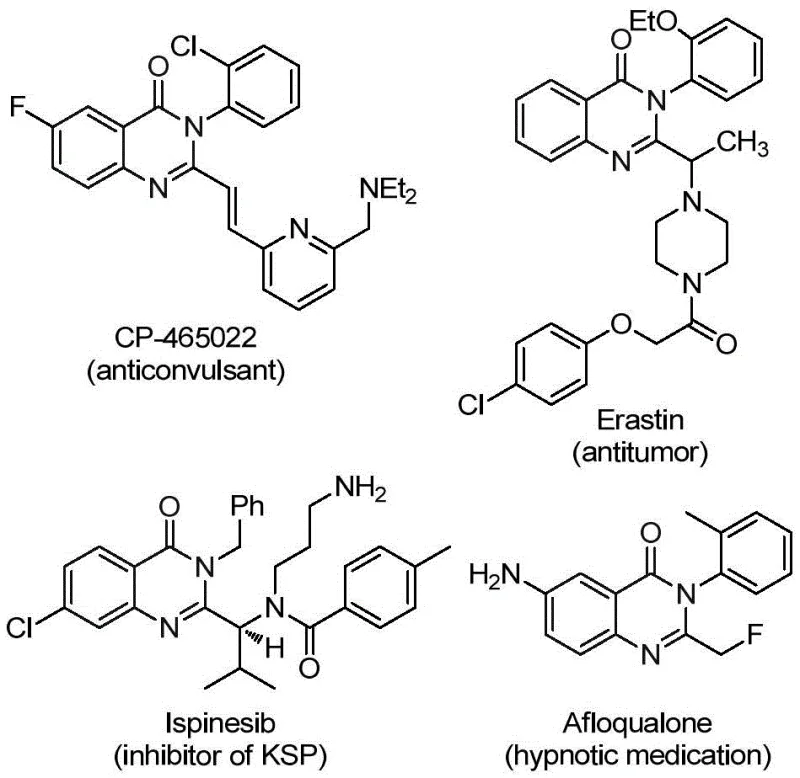 This patent provides a streamlined pathway to access these valuable motifs, offering a distinct advantage over legacy methods that often suffer from harsh conditions or limited substrate tolerance. For R&D directors and procurement managers alike, the ability to source intermediates synthesized via such efficient routes translates directly into accelerated timelines and optimized cost structures for API manufacturing.
This patent provides a streamlined pathway to access these valuable motifs, offering a distinct advantage over legacy methods that often suffer from harsh conditions or limited substrate tolerance. For R&D directors and procurement managers alike, the ability to source intermediates synthesized via such efficient routes translates directly into accelerated timelines and optimized cost structures for API manufacturing.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of 2-trifluoromethyl substituted quinazolinones has been fraught with significant operational and chemical hurdles. Conventional literature reports typically rely on cyclization reactions involving anthranilamides with ethyl trifluoroacetate, trifluoroacetic anhydride, or trifluoroacetic acid under varying conditions. Alternative strategies involve the cyclization of anthranilic esters with unstable trifluoroacetamides or the reaction of isatoic anhydrides with trifluoroacetic anhydride. While these methods can yield the desired products, they are often plagued by severe limitations that hinder their utility in a commercial setting. Common drawbacks include the requirement for harsh reaction conditions that can degrade sensitive functional groups, the necessity for expensive or pre-activated substrates that drive up raw material costs, and generally low yields that compromise overall process efficiency. Furthermore, many traditional protocols exhibit a narrow substrate scope, failing to accommodate diverse electronic environments on the aromatic rings, which restricts the chemical space available for medicinal chemists during lead optimization.
The Novel Approach
In stark contrast to these legacy techniques, the methodology described in patent CN112125856A offers a transformative solution through a transition metal palladium-catalyzed carbonylative tandem reaction. This innovative approach utilizes readily available and inexpensive starting materials, specifically o-iodoaniline and trifluoroethylimidoyl chloride, to construct the quinazolinone core with high efficiency. A pivotal feature of this novel route is the utilization of 1,3,5-tricarboxylate phenol ester (TFBen) as a solid carbon monoxide substitute. This strategic substitution eliminates the need for handling toxic, colorless carbon monoxide gas, thereby drastically improving workplace safety and simplifying reactor requirements. 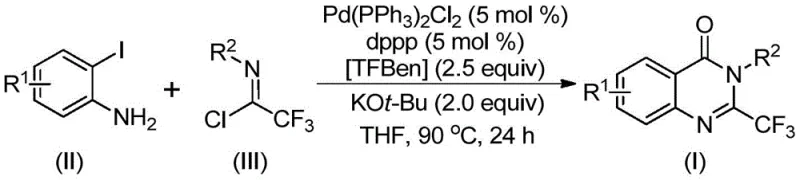 The reaction proceeds smoothly in organic solvents such as tetrahydrofuran at moderate temperatures around 90°C, demonstrating excellent compatibility with a wide range of substituents. This broad substrate applicability ensures that cost reduction in pharmaceutical intermediate manufacturing is achievable without sacrificing chemical diversity or purity standards.
The reaction proceeds smoothly in organic solvents such as tetrahydrofuran at moderate temperatures around 90°C, demonstrating excellent compatibility with a wide range of substituents. This broad substrate applicability ensures that cost reduction in pharmaceutical intermediate manufacturing is achievable without sacrificing chemical diversity or purity standards.
Mechanistic Insights into Palladium-Catalyzed Carbonylative Cyclization
To fully appreciate the robustness of this synthetic route, one must delve into the mechanistic intricacies that govern the transformation. The reaction is believed to initiate with an intermolecular carbon-nitrogen bond coupling promoted by the base, potassium tert-butoxide, generating a trifluoroacetamidine derivative intermediate. Subsequently, the palladium catalyst, typically bis(triphenylphosphine)palladium(II) dichloride coordinated with a diphosphine ligand like dppp, undergoes oxidative addition into the carbon-iodine bond of the o-iodoaniline moiety. This step forms a crucial divalent palladium intermediate that sets the stage for the subsequent carbonylation event. Under the heating conditions employed, the solid CO surrogate TFBen decomposes to release carbon monoxide in situ. This generated carbon monoxide then inserts into the carbon-palladium bond, creating an acyl-palladium species. The presence of the base further facilitates the formation of a palladium-nitrogen bond, leading to a seven-membered ring palladium intermediate. The cycle concludes with a reductive elimination step that releases the final 2-trifluoromethyl substituted quinazolinone derivative and regenerates the active palladium catalyst for another turnover.
From a quality control perspective, understanding this mechanism is vital for impurity management. The use of a well-defined catalytic cycle minimizes side reactions such as homocoupling of the aryl iodide or hydrodehalogenation, which are common pitfalls in palladium chemistry. The specific choice of ligands and the controlled release of carbon monoxide from the solid surrogate ensure that the carbonylation step occurs with high selectivity. This precision is critical for maintaining a clean impurity profile, a key metric for any reliable pharmaceutical intermediate supplier. The method's tolerance for various substituents, including halogens like fluorine, chlorine, and bromine, as well as electron-withdrawing nitro groups and bulky naphthyl systems, underscores its versatility. 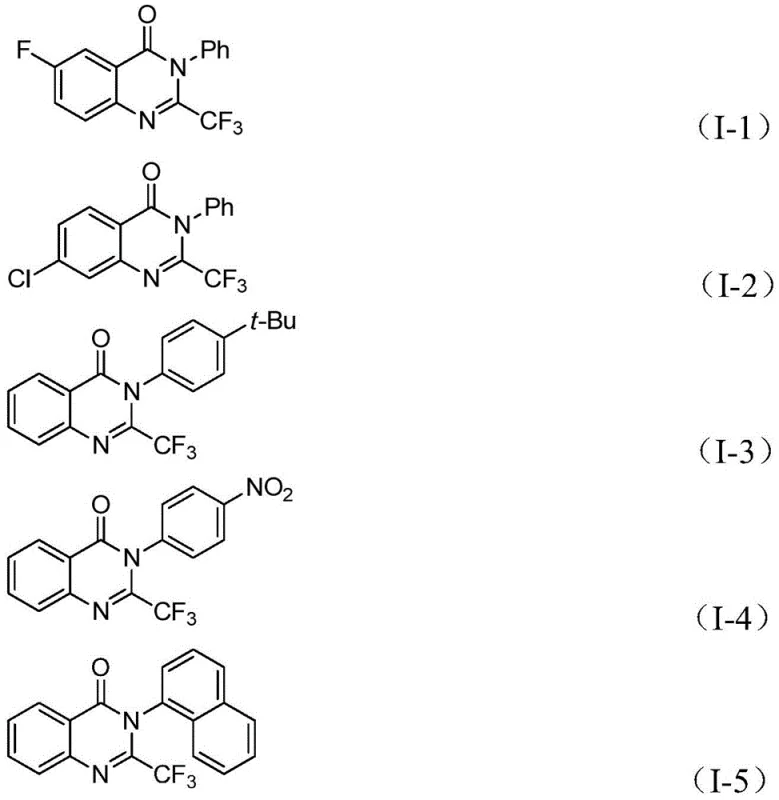 Such structural diversity allows for the tailored synthesis of complex molecules required for structure-activity relationship (SAR) studies, ensuring that the commercial scale-up of complex pharmaceutical intermediates remains feasible and efficient.
Such structural diversity allows for the tailored synthesis of complex molecules required for structure-activity relationship (SAR) studies, ensuring that the commercial scale-up of complex pharmaceutical intermediates remains feasible and efficient.
How to Synthesize 2-Trifluoromethyl Quinazolinone Efficiently
The practical execution of this synthesis is designed for reproducibility and ease of operation, making it highly attractive for process development teams. The protocol involves charging a reaction vessel with the palladium catalyst, ligand, base, solid CO source, and the two primary coupling partners in an aprotic solvent. The mixture is then heated to facilitate the tandem cyclization. This streamlined workflow reduces the number of unit operations required compared to multi-step traditional syntheses, directly impacting the overall production timeline and resource allocation.
- Combine palladium catalyst, dppp ligand, potassium tert-butoxide, TFBen (solid CO source), trifluoroethylimidoyl chloride, and o-iodoaniline in an organic solvent like THF.
- Heat the reaction mixture to 90°C and maintain stirring for 16 to 30 hours to allow the carbonylative cyclization to proceed.
- Upon completion, filter the mixture, mix with silica gel, and purify via column chromatography to isolate the final 2-trifluoromethyl-substituted quinazolinone derivative.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this patented methodology offers tangible strategic benefits that extend beyond mere chemical yield. The shift towards safer, more efficient synthetic routes is a primary driver for reducing operational risks and ensuring supply continuity. By replacing hazardous gaseous reagents with stable solid surrogates, the process mitigates the risks associated with gas storage and handling, which can often be a bottleneck in regulatory compliance and facility licensing. This inherent safety feature translates into a more resilient supply chain, as it reduces the dependency on specialized infrastructure that might be prone to disruptions.
- Cost Reduction in Manufacturing: The economic viability of this process is underpinned by the use of cheap and commercially available starting materials. O-iodoanilines and trifluoroethylimidoyl chlorides are accessible commodities, avoiding the need for custom-synthesized, high-cost precursors often required in older methods. Furthermore, the elimination of toxic gas handling equipment reduces capital expenditure and maintenance costs. The high conversion rates observed with this method mean that less raw material is wasted, optimizing the atom economy and driving down the cost per kilogram of the final intermediate. This efficiency is crucial for maintaining competitive pricing in the global market for high-purity pharmaceutical intermediates.
- Enhanced Supply Chain Reliability: The robustness of the reaction conditions contributes significantly to supply reliability. The method operates effectively in common organic solvents like tetrahydrofuran, which are widely available and easy to source globally. The tolerance for a broad range of functional groups means that supply chain disruptions affecting specific substituted anilines can often be mitigated by switching to alternative analogs without needing to re-optimize the entire process. This flexibility ensures that production schedules can be maintained even when facing raw material volatility, providing a stable flow of materials for downstream API synthesis.
- Scalability and Environmental Compliance: Scaling chemical processes from the bench to the plant floor often introduces new challenges, particularly regarding heat transfer and mass transfer with gaseous reagents. By utilizing a solid CO source, this method simplifies the engineering requirements for scale-up, as it avoids the complexities of gas-liquid mass transfer limitations. Additionally, the reduced use of hazardous reagents aligns with increasingly stringent environmental regulations. The simpler post-treatment process, involving filtration and standard chromatography, minimizes waste generation and solvent consumption, supporting sustainable manufacturing practices and reducing the environmental footprint of the production facility.
Frequently Asked Questions (FAQ)
Understanding the technical details of a new synthetic method is crucial for making informed sourcing decisions. The following questions address common inquiries regarding the safety, scope, and practical application of this palladium-catalyzed carbonylation technology. These insights are derived directly from the experimental data and specifications outlined in the patent documentation, ensuring accuracy and relevance for technical stakeholders. Addressing these points proactively helps in aligning expectations between suppliers and manufacturing partners.
Q: How does this method improve safety compared to traditional quinazolinone synthesis?
A: Traditional methods often require handling toxic carbon monoxide gas directly. This patented process utilizes TFBen (1,3,5-tricarboxylate phenol ester) as a solid carbon monoxide substitute, significantly enhancing operational safety and eliminating the need for specialized gas handling infrastructure.
Q: What is the substrate scope for this palladium-catalyzed reaction?
A: The method demonstrates excellent compatibility with various substituents. It tolerates electron-withdrawing and electron-donating groups on the aryl ring, including halogens (F, Cl, Br), alkyl groups, and nitro groups, allowing for the synthesis of diverse derivatives suitable for SAR studies.
Q: Is the catalyst loading cost-effective for large-scale production?
A: Yes, the process utilizes a relatively low loading of palladium catalyst (typically around 5 mol%) combined with inexpensive ligands and readily available starting materials like o-iodoaniline and trifluoroethylimidoyl chloride, making it economically viable for industrial scaling.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 2-Trifluoromethyl Quinazolinone Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical role that advanced synthetic methodologies play in accelerating drug development. Our team of expert chemists is well-versed in implementing cutting-edge technologies like the palladium-catalyzed carbonylation described in patent CN112125856A. We possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your project needs are met with precision and reliability. Our commitment to quality is backed by stringent purity specifications and rigorous QC labs that utilize state-of-the-art analytical instrumentation to verify every batch. Whether you require custom synthesis for early-stage research or large-scale manufacturing for clinical trials, our infrastructure is designed to support your growth.
We invite you to collaborate with us to leverage these technological advantages for your specific projects. Our technical procurement team is ready to provide a Customized Cost-Saving Analysis tailored to your volume requirements and timeline. By partnering with us, you gain access to specific COA data and comprehensive route feasibility assessments that will empower your decision-making process. Contact us today to discuss how we can support your supply chain with high-quality 2-trifluoromethyl quinazolinone derivatives and other complex pharmaceutical intermediates.