Scalable Iodine-Promoted Synthesis of 3,4,5-Trisubstituted 1,2,4-Triazoles for Commercial API Production
Introduction to Advanced Triazole Synthesis Technology
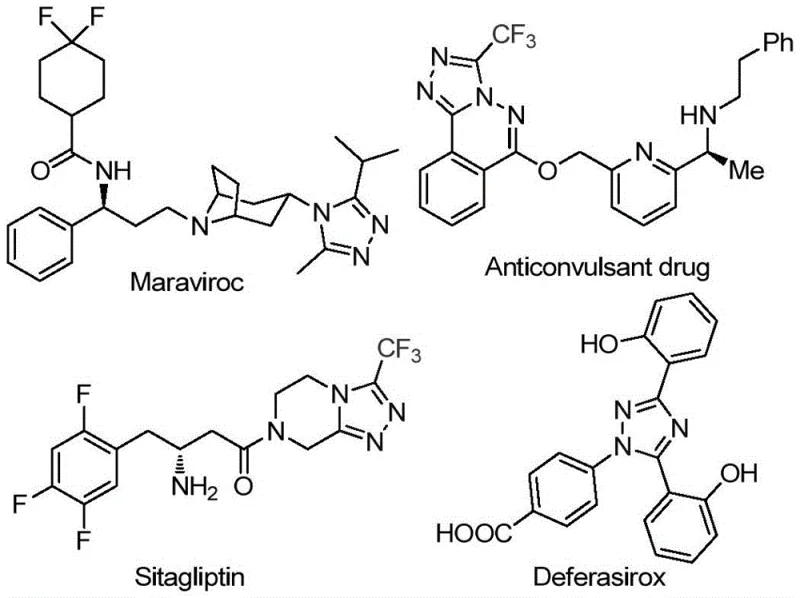
The pharmaceutical industry continuously demands robust and scalable synthetic routes for nitrogen-containing heterocycles, particularly 1,2,4-triazoles, which serve as critical scaffolds in numerous bioactive molecules. As highlighted in recent intellectual property developments, specifically patent CN113105402B, a significant breakthrough has been achieved in the preparation of 3,4,5-trisubstituted 1,2,4-triazole compounds. This novel methodology addresses long-standing challenges in heterocyclic chemistry by offering a metal-free, operationally simple, and highly efficient pathway. The relevance of this chemical class cannot be overstated, as evidenced by its presence in major therapeutic agents such as Maraviroc, Sitagliptin, and Deferasirox, structures of which are depicted below to illustrate the pharmacological significance of this core motif.
For R&D directors and process chemists, the ability to introduce trifluoromethyl groups alongside acyl functionalities into the triazole ring represents a substantial value add, enhancing metabolic stability and lipophilicity. The disclosed technology leverages a unique tandem reaction sequence involving iodine-promoted oxidation and subsequent cyclization. This approach not only simplifies the synthetic workflow but also aligns with modern green chemistry principles by avoiding hazardous reagents. As a reliable pharmaceutical intermediate supplier, understanding these mechanistic nuances is essential for evaluating the feasibility of integrating such intermediates into complex API manufacturing pipelines.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the construction of polysubstituted 1,2,4-triazole rings has relied heavily on transition metal catalysis or harsh reaction conditions that pose significant hurdles for industrial scale-up. Traditional protocols often necessitate the use of expensive copper or palladium catalysts, which introduce complications regarding residual metal removal—a critical quality attribute for any pharmaceutical intermediate intended for human consumption. Furthermore, many existing methods require strictly anhydrous and oxygen-free environments, demanding specialized equipment and inert gas manifolds that increase capital expenditure and operational complexity. The sensitivity of reagents in conventional pathways often leads to inconsistent batch-to-batch reproducibility, creating supply chain vulnerabilities for procurement managers seeking consistent quality.
The Novel Approach
In stark contrast, the methodology outlined in the patent data introduces a paradigm shift by utilizing elemental iodine and dimethyl sulfoxide (DMSO) as the primary drivers of reactivity. This metal-free strategy eliminates the burden of heavy metal scavenging steps, thereby streamlining the downstream purification process. The reaction proceeds through a clever cascade where aryl ethyl ketones undergo iodination and Kornblum oxidation in situ to generate reactive dicarbonyl species, which then condense with trifluoroethylimide hydrazides. This one-pot transformation significantly reduces solvent usage and waste generation. By operating under ambient atmospheric conditions without the need for rigorous exclusion of moisture or oxygen, this novel approach drastically lowers the barrier to entry for commercial production, offering a distinct advantage in cost reduction in pharmaceutical intermediate manufacturing.
Mechanistic Insights into Iodine-Promoted Cyclization
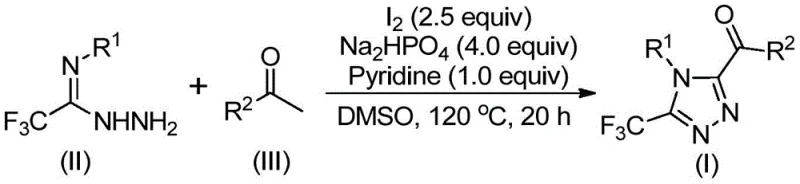
The core of this technological advancement lies in the dual role of iodine as both an oxidant and a cyclization promoter. The reaction initiates with the activation of the aryl ethyl ketone by iodine in DMSO, facilitating an alpha-iodination followed by a Kornblum oxidation to yield an alpha-dicarbonyl intermediate. This electrophilic species is then poised for nucleophilic attack by the hydrazide nitrogen of the trifluoroethylimide hydrazide. The subsequent dehydration leads to a hydrazone intermediate, which undergoes an intramolecular cyclization driven by the basic environment provided by pyridine and sodium dihydrogen phosphate. This mechanistic pathway ensures high regioselectivity, favoring the formation of the desired 3,4,5-trisubstituted isomer over potential byproducts.
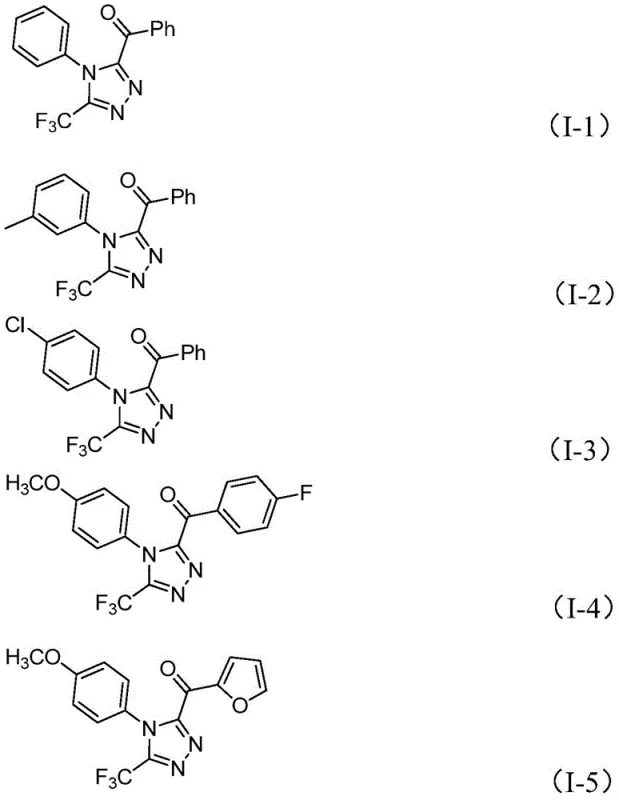
From an impurity control perspective, the use of mild inorganic bases like sodium dihydrogen phosphate helps maintain a buffered pH that minimizes side reactions such as over-oxidation or polymerization of the sensitive intermediates. The tolerance of the system towards various functional groups—including halogens, alkoxy groups, and trifluoromethyl substituents on the aromatic rings—demonstrates the robustness of the catalytic cycle. This broad substrate scope allows medicinal chemists to explore diverse chemical space without redesigning the synthetic route for each new analog. The structural diversity achievable is exemplified by the range of products shown below, confirming the versatility of this platform for generating high-purity pharmaceutical intermediates with tailored physicochemical properties.
How to Synthesize 3,4,5-Trisubstituted 1,2,4-Triazoles Efficiently
Implementing this synthesis requires precise control over stoichiometry and thermal profiles to maximize yield and purity. The process begins with the dissolution of the aryl ethyl ketone and a portion of elemental iodine in DMSO, followed by a controlled heating phase to generate the oxidized intermediate. Subsequent addition of the hydrazide component along with the base system triggers the cyclization event. Detailed standard operating procedures regarding exact molar ratios, such as the preferred 1:2:4:1:2.5 ratio of hydrazide to ketone to phosphate to pyridine to iodine, are critical for reproducibility. For a comprehensive breakdown of the standardized synthesis steps, please refer to the technical guide below.
- Combine aryl ethyl ketone and elemental iodine in dimethyl sulfoxide (DMSO) and heat to 90-110°C for 4-6 hours to initiate Kornblum oxidation.
- Add additional iodine, sodium dihydrogen phosphate, pyridine, and trifluoroethylimide hydrazide to the reaction mixture.
- Heat the mixture to 110-130°C for 12-20 hours to complete the cyclization, followed by filtration and column chromatography purification.
Commercial Advantages for Procurement and Supply Chain Teams
For supply chain leaders and procurement specialists, the adoption of this iodine-promoted synthesis offers tangible strategic benefits beyond mere chemical elegance. The elimination of precious metal catalysts directly translates to a reduction in raw material costs and simplifies the regulatory dossier by removing the need for extensive heavy metal testing. Moreover, the reliance on commodity chemicals like iodine, DMSO, and commercially available acetophenones ensures a stable and resilient supply base, mitigating risks associated with sourcing exotic reagents. The operational simplicity of the process, which does not demand specialized glovebox techniques or cryogenic cooling, allows for deployment in standard multipurpose reactors, enhancing manufacturing flexibility.
- Cost Reduction in Manufacturing: The economic impact of switching to this metal-free protocol is profound. By removing the requirement for expensive transition metal catalysts and the associated ligands, the direct material cost per kilogram of product is significantly lowered. Additionally, the simplified workup procedure, which avoids complex metal scavenging resins or extensive aqueous extractions, reduces labor hours and solvent consumption. This streamlined workflow results in substantial cost savings that can be passed down the supply chain, improving the overall margin profile for the final API.
- Enhanced Supply Chain Reliability: The robustness of the reaction conditions contributes to higher process reliability and shorter lead times. Since the reaction tolerates ambient atmosphere and does not require rigorously dried solvents, the risk of batch failure due to environmental factors is minimized. The starting materials, including various substituted acetophenones and hydrazides, are widely available from global chemical suppliers, ensuring continuity of supply even during market fluctuations. This stability is crucial for maintaining just-in-time inventory levels and meeting tight production schedules.
- Scalability and Environmental Compliance: Scaling this process from gram to multi-kilogram quantities is straightforward due to the exothermic nature of the reaction being manageable within standard thermal limits. The use of DMSO, a high-boiling polar aprotic solvent, facilitates high concentration reactions, reducing the reactor volume footprint. Furthermore, the absence of toxic heavy metals simplifies waste stream treatment and disposal, aligning with increasingly stringent environmental regulations and corporate sustainability goals. This eco-friendly profile enhances the long-term viability of the manufacturing process.
Frequently Asked Questions (FAQ)
To assist technical teams in evaluating this technology for their specific applications, we have compiled answers to common inquiries regarding the reaction parameters and scope. These insights are derived directly from the experimental data and optimization studies presented in the patent literature. Understanding these nuances helps in assessing the fit for your specific project requirements and anticipating potential scale-up considerations.
Q: Does this synthesis method require expensive transition metal catalysts?
A: No, the method described in patent CN113105402B utilizes elemental iodine as a non-metallic promoter, eliminating the need for costly and toxic heavy metal catalysts often found in traditional triazole synthesis.
Q: What are the optimal reaction conditions for scaling this process?
A: The process operates effectively in DMSO solvent with a two-stage heating profile: an initial oxidation at 90-110°C followed by cyclization at 110-130°C, without requiring strict anhydrous or oxygen-free environments.
Q: Is this method suitable for producing diverse triazole derivatives?
A: Yes, the protocol demonstrates high substrate tolerance, successfully accommodating various substituted aryl groups and heteroaryl groups at both the R1 and R2 positions, making it highly versatile for library synthesis.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 3,4,5-Trisubstituted 1,2,4-Triazole Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical role that advanced heterocyclic intermediates play in accelerating drug discovery and development. Our team of expert process chemists possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that innovative laboratory methods like the one described in CN113105402B can be seamlessly translated into industrial reality. We are committed to delivering products with stringent purity specifications, supported by our rigorous QC labs equipped with state-of-the-art analytical instrumentation to verify identity and assay.
We invite you to collaborate with us to leverage this efficient synthetic route for your next project. Contact our technical procurement team today to request a Customized Cost-Saving Analysis tailored to your volume requirements. We are ready to provide specific COA data and route feasibility assessments to demonstrate how our capabilities can support your supply chain objectives and drive your pipeline forward.